Last update: 29 września 2016


Wprowadzenie
Badania zwykle prowadzone na wczesnym etapie rozwoju klinicznego (badania fazy I i fazy II) mają różne cele. W tych wczesnych badaniach klinicznych przede wszystkim konieczne jest ustalenie, że badany produkt leczniczy jest bezpieczny dla ludzi. Stanowią one również próbę wykazania, że lek jest skuteczny przeciw docelowej chorobie lub stanowi.
W trakcie wczesnego stadium rozwoju klinicznego należy odpowiedzieć na następujące pytania:
- Faza I
- Czy lek jest bezpieczny dla ludzi? Na jakich poziomach? (Tolerancja)
- Jak organizm przetwarza lek? (Farmakokinetyka (PK))
- Jak lek działa w organizmie? (Farmakodynamika (PD))
- Jakie istnieją interakcje? (interakcje lek-lek, interakcje z żywnością i napojami itd.)
- Czy lek jest czynny?
- Faza II
- Czy lek jest bezpieczny dla pacjentów? (Bezpieczeństwo stosowania)
- Jak lek działa w organizmie? (Farmakodynamika (PD))
- Czy lek wydaje się skuteczny u pacjentów? W jakiej dawce/dawkach? (Działanie)
- Jak powinny być zaprojektowane badania potwierdzające? (Punkty końcowe, populacja docelowa, inne przyjmowane leki (towarzyszące) itd.)
Choć prace rozwojowe nad lekiem są zwykle przedstawiane jako szereg etapów następujących po sobie w porządku chronologicznym, badania prowadzone na każdym etapie są zwykle klasyfikowane na podstawie swoich celów. Jak pokazano na poniższym schemacie, badania, które zwykle są częścią wcześniejszego etapu, mogą być przeprowadzone później w ramach procesu rozwoju leku, w miarę jak pojawiające się dane wskazują na potrzebę uzyskania dodatkowych informacji.
Badania z eskalacją dawki metodą jednorazowej/wielorazowej dawki rosnącej (SAD i MAD)
Badania metodą jednorazowej dawki rosnącej (ang. single ascending dose, SAD) i wielorazowej dawki rosnącej (multiple ascending dose, MAD) są zwykle pierwszymi badaniami z udziałem ludzi.
Cele badania
Badania SAD i MAD:
- Ocena bezpieczeństwa i tolerancji
- Ustalenie maksymalnej dawki tolerowanej (ang. maximum tolerated dose, MTD)
- Ocena ogólnych właściwości farmakokinetycznych (PK)
- Zbadanie sposobu osiągania stabilnego poziomu leku w organizmie w miarę upływu czasu. Warunki te są nazywane parametrami stanu stacjonarnego (zależność kumulacji leku od czasu)
- Przeprowadzenie wstępnego badania wydalania leku z organizmu (rozpoznanie metabolitów)
Projekt badania
Dawka początkowa w badaniach SAD/MAD jest określana na podstawie wyników nieklinicznych badań toksykologicznych. Dawka jest następnie zwiększana zgodnie z harmonogramami eskalacji określonymi w wytycznych wydanych przez organy regulacyjne. Badania są przerywane zgodnie z zasadami przerywania badania, m.in. po stwierdzeniu toksyczności i braku toksyczności (przy maksymalnie zwiększonej ekspozycji, maksymalnie zwiększonej farmakodynamice itp.).
Badania bilansu masy: wchłanianie, dystrybucja, metabolizm, wydalanie (ADME)
Cele badania
Celem badań ADME (ang. absorption, distribution, metabolism, excretion) jest zrozumienie i scharakteryzowanie profilu farmakokinetycznego (PK) badanego produktu leczniczego, tj. tego, co się dzieje z lekiem w organizmie. W badaniach tych oceniany jest sposób wchłaniania leku przez organizm, sposób jego dystrybucji w organizmie, sposób metabolizowania leku przez organizm oraz sposób wydalania leku przez organizm (zob. rycina poniżej).
-
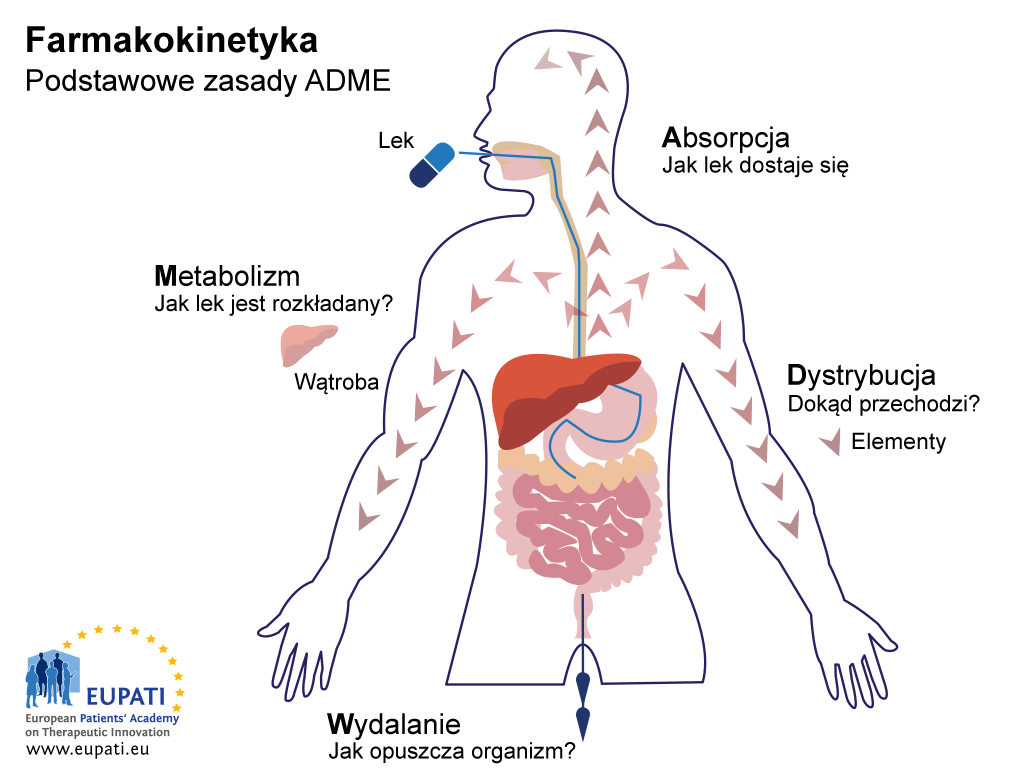
- Podstawy farmakokinetyki — nauki badającej oddziaływanie organizmu na lek — przedstawia akronim ADME.
Projekt badania
Badania ADME są zwykle prowadzone z wykorzystaniem pojedynczej dawki leku u niewielkiej liczby (zwykle u czterech do sześciu) zdrowych mężczyzn (aby wykluczyć potencjalne szkody u kobiet mogących zajść w ciążę) przy zastosowaniu zamierzonego sposobu podawania. Badania te są zwykle związane z fazą I prac rozwojowych, ale mogą być prowadzone przez cały okres rozwoju produktu leczniczego.
Badana ADME dostarczają też informacji o biodostępności leku, tj. ułamku podanej dawki składnika czynnego (ang. active pharmaceutical ingredient, API) docierającym do krwiobiegu.
Badania biodostępności (BA) i biorównoważności (BE)
Cele badania
Badania biodostępności mają na celu ocenę szybkości i stopnia wchłaniania leku. Oceniane jest stężenie składnika czynnego (API) w krwiobiegu w miarę upływu czasu.
Leki podawane różnymi drogami mają różne profile biodostępności. Na przykład leki podawane bezpośrednio do krwiobiegu we wstrzyknięciu dożylnym (IV) mają bezpośrednio po podaniu biodostępność na poziomie 100%, natomiast leki podawane doustnie stają się biodostępne, dopiero kiedy dotrą do krwiobiegu po ich wchłonięciu.
-
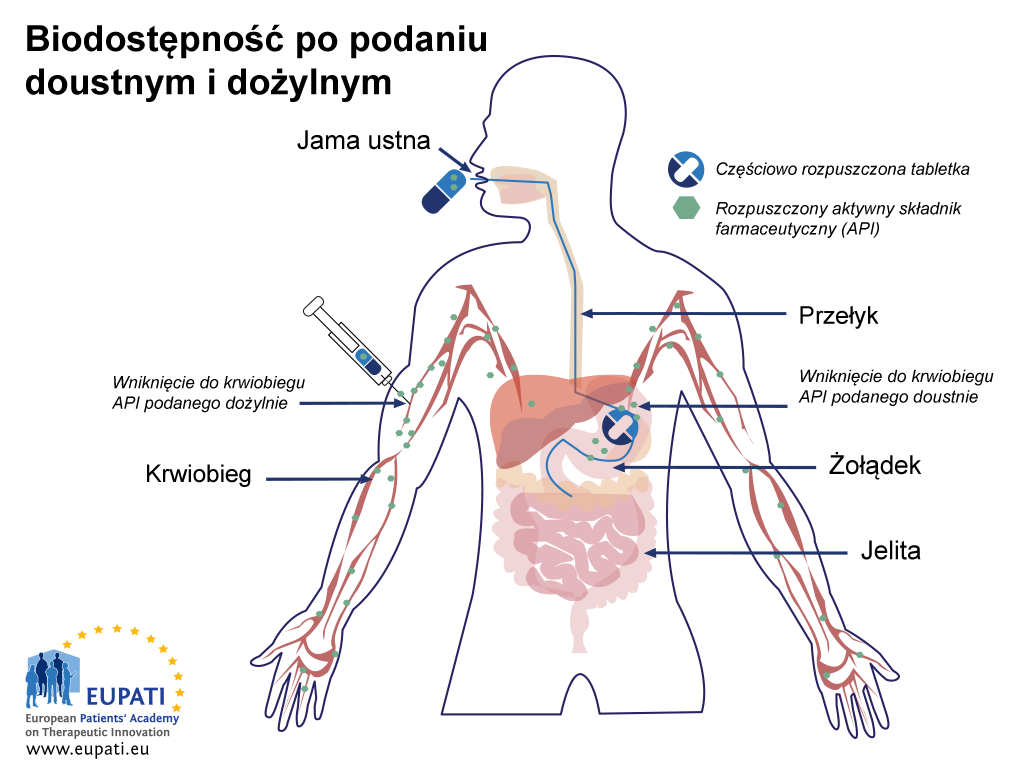
- Schemat przedstawiający różnicę wchłaniania w przypadku kapusłki podanej doustnie a wstrzyknięciem bezpośrednio do krwiobiegu (wstrzyknięcie dożylne). Z żołądka kapsułka przechodzi do jelita cienkiego, gdzie ma miejsce kolejny etap wchłaniania.
W badaniach biodostępności oceniana jest szybkość wchłaniania leku poprzez pomiar maksymalnego stężenia leku (Cmax) w krwiobiegu oraz czasu, w jakim osiągane jest to maksymalne stężenie (Tmax). Obszar pod krzywą (AUC) przedstawia łączną ekspozycję organizmu na działanie leku i jest wykorzystywany w badaniu stopnia wchłaniania.
-
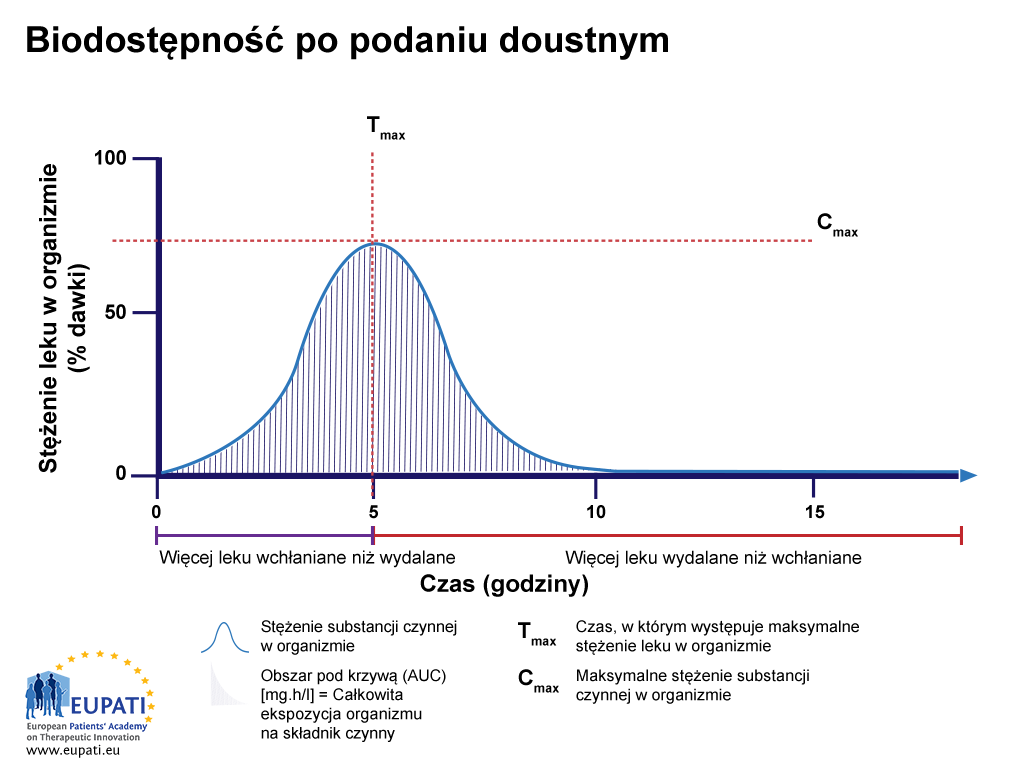
- Procent substancji aktywnej po połknięciu tabletki, badany przez 15 godzin. Obszar AUC jest zacieniony. Tmaks. to czas, po którym lek osiąga najwyższe stężenie w krwiobiegu, natomiast Cmaks. to maksymalne stężenie leku oznaczone w krwiobiegu.
Projekt badania
Zwykle są to randomizowane badania z udziałem zdrowych osób, prowadzone w układzie naprzemiennym po podaniu dawki pojedynczej. Badacze mierzą stężenie leku i jego głównych czynnych metabolitów we krwi i osoczu uczestników.
Badania biorównoważności
W badaniach biorównoważności oceniana jest relacja pomiędzy dwiema różnymi postaciami farmaceutycznymi leku. Tu również oceniana jest szybkość i stopień wchłaniania leku, ale parametry te są porównywane z szybkością i stopniem wchłaniania innego leku lub innej postaci farmaceutycznej tego samego leku (referencyjna postać farmaceutyczna / referencyjny lek). Badania biorównoważności są stosowane w celu porównywania leków generycznych z lekami referencyjnymi. Zanim lek zostanie uznany za biorównoważny z innym lekiem, musi spełnić określone kryteria.
Badania wpływu pokarmów
Cele badania
W badaniach wpływu pokarmów oceniany jest wpływ pokarmów na szybkość i stopień wchłaniania leku oraz jego biodostępność w przypadku określonej postaci farmaceutycznej. Informacje uzyskane w badaniu wpływu pokarmów są ważne z punktu widzenia instrukcji podawania leku zamieszczanej w ulotce dla pacjenta, w której znajduje się informacja, czy lek powinien być przyjmowany na czczo czy z posiłkiem.
Projekt badania
Zwykle są to badania prowadzone w układzie naprzemiennym po podaniu dawki pojedynczej, porównujące grupę uczestników będących na czczo z grupą uczestników po wysokotłuszczowym, wysokokalorycznym posiłku. Badanie jest zwykle prowadzone w dwóch sekwencjach, z udziałem zdrowych uczestników przyjmujących lek o najwyższej przewidywanej mocy.
Badania w zaburzeniach czynności nerek
Cele badania
Celem badań w zaburzeniach czynności nerek jest ocena leku u osób o różnym poziomie zaburzeń czynności nerek. W badaniach tych gromadzone są informacje na temat wpływu zaburzeń czynności nerek na wydalanie leku z organizmu oraz na temat zalecanego dawkowania u pacjentów na różnych etapach zaburzeń czynności nerek.
Projekt badania
Badania w zaburzeniach czynności nerek są prowadzone z udziałem zdrowych ochotników płci męskiej i żeńskiej (około sześciu osób na grupę), w grupach równoległych po podaniu dawki pojedynczej. Grupy te są stratyfikowane na podstawie biomarkerów czynności nerek.
Badania w zaburzeniach czynności wątroby
Cele badania
Celem badań w zaburzeniach czynności wątroby jest ocena leku u osób o różnym poziomie zaburzeń czynności wątroby. W badaniach tych oceniany jest wpływ zaburzeń czynności wątroby na farmakokinetykę leku i jego metabolitów. Przyczyniają się one do określenia zalecanego dawkowania u pacjentów na różnych etapach zaburzeń czynności wątroby z uwzględnieniem skuteczności i/lub bezpieczeństwa.
Projekt badania
Jeśli wyniki parametrów farmakokinetyki uzyskane w poprzednich badaniach są liniowe i niezależne od czasu, badania w zaburzeniach czynności wątroby są zwykle prowadzone jako badania w grupach równoległych z udziałem zdrowych ochotników płci męskiej i żeńskiej (około ośmiu osób), u których występują zaburzenia czynności wątroby o różnym nasileniu. Grupy badane są stratyfikowane na podstawie standardowych klasyfikacji zaburzeń czynności wątroby.
Jeśli lek jest metabolizowany przez enzym, który występuje wyłącznie z powodu zmienności genetycznej, uczestnicy powinni być oceniani na podstawie statusu swojego genotypu.
Badania interakcji lekowych (interakcje leku z lekiem, DDI)
Cele badania
Badania interakcji lekowych (powszechnie znane jako badania interakcji leku z lekiem, ang. drug-drug interaction (DDI)) mają na celu ocenę wpływu równoczesnego podawania leków na farmakokinetykę badanego produktu leczniczego, jak również wpływu badanego produktu leczniczego na farmakokinetykę równocześnie podawanych leków.
Projekt badania
Te badania są prowadzone w oparciu o wyniki wcześniej przeprowadzonych badań w warunkach in vitro; najlepiej, aby były prowadzone w układzie naprzemiennym. Badania interakcji lekowych są prowadzone z udziałem zdrowych ochotników lub z udziałem pacjentów, jeśli wskazana jest ocena farmakodynamicznych punktów końcowych w przypadku zbyt toksycznych leków (na przykład leków przeciwnowotworowych).
Zwykle badania te prowadzone są w układzie naprzemiennym. Dawkowanie, odstępy między dawkami, liczba dawek, drogi podania oraz harmonogram podawania jednocześnie przyjmowanych leków powinny być tak zaplanowane, aby maksymalnie zwiększyć możliwość wykrycia interakcji, i powinny imitować warunki kliniczne. Wielkość interakcji (hamowanie/indukowanie) jest klasyfikowana na podstawie zmiany wchłaniania jednego z badanych produktów leczniczych obliczanej jako obszar pod krzywą (AUC) dla substancji.
Dokładne badanie QT (TQT)
Cele badania
Odstęp QT jest miarą rytmu serca. Odstęp QT może być mierzony za pomocą elektrokardiogramu (EKG) i wykorzystany jako (niedoskonały) biomarker do oceny ryzyka wywołania arytmii przez dany lek. Podczas badania EKG mierzona jest elektryczna aktywność serca prezentowana w postaci fal oznaczonych jako „P”, „Q”, „R”, „S” i „T”. Odstęp QT jest mierzony między początkiem fali Q a końcem fali T.
Projekt badania
Badania TQT są prowadzone w warunkach in vivo dla wszystkich nowych cząsteczek (ang. new molecular entity, NME). Muszą się one odbyć przed badaniami fazy III niezależnie od wyników badań w warunkach in vitro czy badań nieklinicznych.
Zwykle badania TQT są prowadzone z udziałem zdrowych osób po podaniu dawki pojedynczej w układzie naprzemiennym. Badacze oceniają terapeutyczne i supraterapeutyczne (większe niż terapeutyczne) dawki leku w porównaniu z kontrolą dodatnią (np. z popularnym antybiotykiem takim jak moksyfloksacyna) i kontrolą ujemną (placebo).
A2-5.03.4-V1.1