Last update: 29 september 2016


Inledning
De studier som är vanliga under tidig klinisk utveckling (fas I och fas II) har en rad olika mål.De tidiga kliniska prövningarna måste framför allt fastställa att ett prövningsläkemedel är säkert för människor.De försöker också visa att läkemedlet är effektivt mot den avsedda sjukdomen eller det avsedda tillståndet.
Följande nyckelfrågor måste besvaras under den tidiga kliniska utvecklingen:
- Fas I
- Är läkemedlet säkert för människor?Vid vilka nivåer?(tolerans)
- Vad gör kroppen med läkemedlet?(farmakokinetik (PK))
- Vad gör läkemedlet med kroppen?(farmakodynamik (PD))
- Vilka interaktioner förekommer?(interaktioner med andra läkemedel, mat, dryck osv.)
- Är läkemedlet aktivt?
- Fas II
- Är läkemedlet säkert för patienter?(säkerhet)
- Vad gör läkemedlet med kroppen?(farmakodynamik (PD))
- Verkar läkemedlet fungera på patienter?I vilken eller vilka doser?(effekt)
- Hur ska bekräftande prövningar utformas?(utfall, målpopulation, andra läkemedel som tas (samtidigt) osv.)
Även om läkemedelsutveckling vanligtvis utgörs av en serie faser i kronologisk ordning klassificeras de studier som utförs inom varje fas i regel efter respektive mål.Som diagrammet nedan visar kan studier som vanligtvis ingår i en tidigare fas utföras senare i läkemedelsutvecklingsprocessen allt eftersom framkomna data visar på behovet av mer information.
Doseskaleringsstudier med engångsdosering/upprepad dosering (SAD och MAD)
Doseskaleringsstudier med engångsdosering (SAD) och upprepad dosering (MAD) är i regel de första studierna på människa (”first-in-human”).
Studiemål
SAD- och MAD-studier:
- Undersöka säkerhet och tolerabilitet
- Identifiera en maximal tolererad dos (MTD)
- Undersöka allmänna farmakokinetiska (PK) egenskaper
- Undersöka hur man uppnår en stabil nivå av läkemedlet i kroppen över tid. Dessa tillstånd är kända som steady-state-parametrar (ackumulering, tidsberoende).
- Utföra preliminär utforskning av läkemedlets utsöndring från kroppen (identifiera metabolit(er))
Studiens utformning
Startdosen i SAD/MAD-studier fastställs utifrån resultaten från icke-kliniska toxikologiska studier.Dosen ökas sedan enligt eskaleringsscheman som har dokumenterats i regulatoriska riktlinjer.Studierna avbryts enligt stoppregler som innefattar toxicitet och obefintlig toxicitet (maximerad exponering, maximerad farmakodynamik osv.).
Studier av ”massbalans”:Absorption, Distribution, Metabolism, Utsöndring (ADME)
Studiemål
Målet med ADME-studierna är att förstå och karakterisera prövningsläkemedlets farmakokinetiska (PK) profil,det vill säga vad kroppen gör med läkemedlet.De här studierna undersöker på vilket sätt kroppen absorberar läkemedlet, hur läkemedlet distribueras i kroppen, hur kroppen metaboliserar läkemedlet och hur kroppen utsöndrar läkemedlet (se bilden nedan).
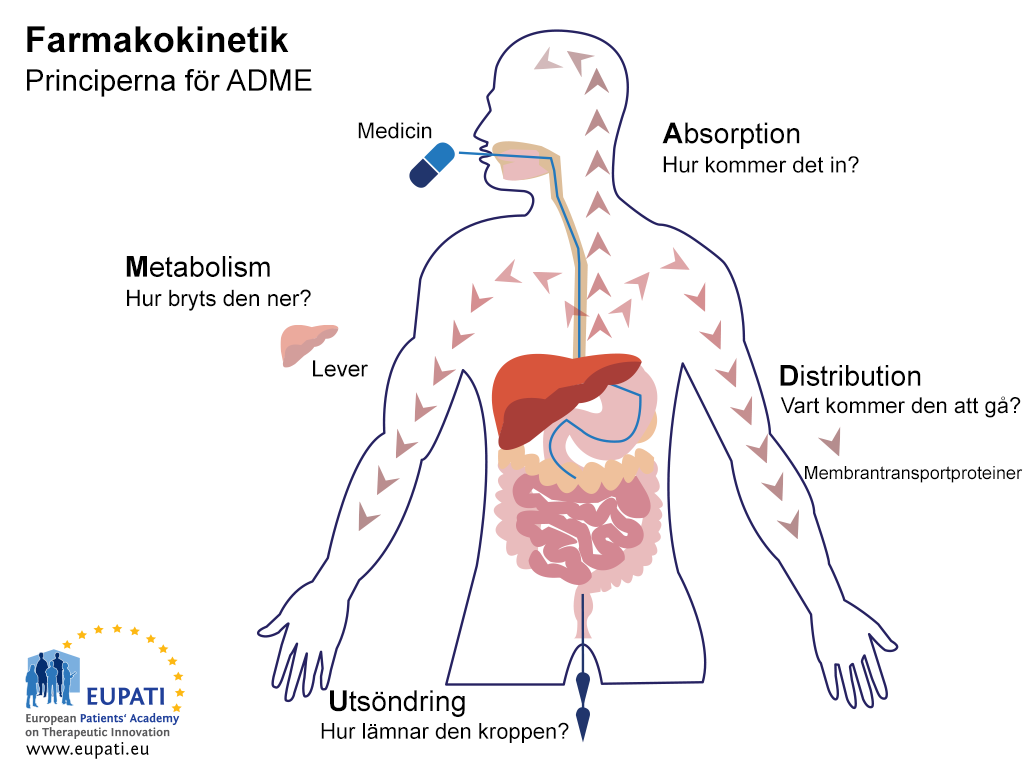
De viktigaste principerna för Farmakokinetik – studiet av effekten kroppen har på ett läkemedel – representeras av akronymen ADME. (Avsiktsförklaring: Den här bilden översattes med hjälp av pålitliga AI-assisterade översättningsverktyg med bevisad noggrannhet och bred flerspråkig kompetens.)
Studiens utformning
ADME-studier utförs i regel med en engångsdos av läkemedlet hos ett litet antal (vanligen fyra till sex) friska män (för att utesluta eventuell risk för fertila kvinnor) via det avsedda administreringssättet.De här studierna är oftast knutna till fas I i utvecklingen, men de kan utföras under hela utvecklingen av ett läkemedel.
ADME-studier ger också information om biotillgänglighet, det vill säga andelen av en administrerad dos av en aktiv substans (API) som når blodomloppet.
Studier av biotillgänglighet (BA) och bioekvivalens (BE)
Studiemål
Biotillgänglighetsstudier utvärderar hastigheten och omfattningen av ett läkemedels absorption.De undersöker koncentrationen av den aktiva substansen (API) i blodomloppet över tid.
Läkemedel med olika administreringssätt har olika biotillgänglighetsprofiler.Till exempel har läkemedel som administreras direkt i blodomloppet via intravenös injektion (IV) 100 % biotillgänglighet i samma ögonblick som de administreras, medan läkemedel som administreras oralt inte blir biotillgängliga förrän läkemedlet når blodomloppet efter absorption.
Biotillgänglighetsstudier bedömer ett läkemedels absorptionshastighet genom att mäta den maximala koncentrationen av läkemedlet (Cmax) i blodomloppet, och tidpunkten då den maximala koncentrationen uppmättes (Tmax).Arean under kurvan (AUC) representerar kroppens totala exponering för läkemedlet och används för att studera absorptionens omfattning.
Studiens utformning
Dessa studier utförs i regel som randomiserade crossover-studier med engångsdos hos friska deltagare.Forskarna mäter koncentrationen av läkemedlet och dess huvudsakliga aktiva metaboliter i deltagarnas blod och plasma.
Bioekvivalensstudier
Bioekvivalensstudier undersöker förhållandet mellan två olika beredningar.De studerar också ett läkemedels absorptionshastighet och -omfattning, men de jämför dessa med absorptionshastigheten och -omfattningen för ett annat läkemedel eller en annan beredning av samma läkemedel (referensberedning/-läkemedel).Bioekvivalensstudier används för att jämföra generiska läkemedel med deras referensläkemedel.Det finns fasta kriterier som ett läkemedel måste uppfylla innan det kan anses vara bioekvivalent med ett annat läkemedel.
Studier av livsmedels effekt
Studiemål
Livsmedelseffektstudier utvärderar vilken effekt mat har på hastigheten, omfattningen och biotillgängligheten i ett läkemedels absorption i en given beredning.Informationen från livsmedelseffektstudier är viktig för administreringsanvisningarna som finns i bipacksedeln (PL) och som talar om ifall läkemedlet ska tas på fastande mage eller i samband med måltid.
Studiens utformning
Dessa studier utförs i regel som crossover-studier med engångsdos och jämför två tillstånd:deltagare som har fastat och deltagare som intagit en måltid med högt fett- och kaloriinnehåll.Studien har i regel två sekvenser och utförs med friska deltagare som får den högsta förväntade styrkan av läkemedlet.
Studier av nedsatt njurfunktion
Studiemål
Målet för studier av nedsatt njurfunktion är att utvärdera läkemedlet hos människor med olika njurfunktionsnivåer.De här studierna samlar in information om effekten av nedsatt njurfunktion på kroppens utsöndring av läkemedlet och dosrekommendationer för patienter med olika stadier av nedsatt njurfunktion.
Studiens utformning
Studier av nedsatt njurfunktion utförs som engångsdosstudier med parallella grupper hos friska frivilliga män och kvinnor (runt sex per grupp).Grupperna stratifieras baserat på biomarkörer för njurfunktion.
Studier av nedsatt leverfunktion
Studiemål
Målet för studier av nedsatt leverfunktion är att utvärdera läkemedlet hos människor med olika nivåer av nedsatt leverfunktion.De här studierna undersöker effekten av nedsatt leverfunktion på läkemedlets farmakokinetik och dess metaboliter, och bidrar med dosrekommendationer för patienter med olika stadier av nedsatt leverfunktion av effekt- och/eller säkerhetsskäl.
Studiens utformning
Om de farmakokinetiska resultaten från tidigare studier är linjära och tidsoberoende genomförs studier av nedsatt leverfunktion i regel som studier med parallella grupper hos friska frivilliga män och kvinnor (runt åtta individer) med olika nedsättningsgrad av leverfunktionen.Behandlingsgrupperna stratifieras baserat på standardklassificeringar av nedsatt leverfunktion.
Om läkemedlet metaboliseras av ett enzym som bara förekommer på grund av genetisk variation ska deltagarana utvärderas utifrån genotypstatus.
Studier av läkemedelsinteraktion (Drug-Drug Interaction, DDI)
Studiemål
Läkemedelsinteraktionsstudier (kallas även ”Drug-Drug Interaction” (DDI)) utvärderar effekten av samtidiga läkemedel på prövningsläkemedlets (IMP) farmakokinetik, samt effekten av IMP på de samtidiga läkemedlens farmakokinetik.
Studiens utformning
De här studierna vägleds av tidigare in vitro-resultat. De utförs med fördel i en crossover-prövning.Läkemedelsinteraktionsstudier utförs på friska frivilliga eller på patienter om det är önskvärt att utvärdera farmakodynamiska utfall när läkemedlen är för toxiska (till exempel cancerläkemedel).
De här studierna är i regel utformade som crossover-prövningar.Dos, dosintervall, antal doser, administreringssätt och tidpunkt för samtidig administrering ska vara utformade för maximal möjlighet att upptäcka en interaktion och ska efterlikna den kliniska miljön.Interaktionens omfattning (hämning/induktion) klassificeras av förändringen i absorptionen av ett av prövningsläkemedlen, beräknat som substansens area under kurvan (AUC).
Grundlig QT-studie (TQT)
Studiemål
Ett QT-intervall är ett mått på hjärtrytmen.Ett QT-intervall kan mätas med ett elektrokardiogram (EKG) och kan användas som en (felaktig) biomarkör för att bedöma risken för att ett läkemedel kan orsaka hjärtklappning (arytmi).I ett EKG mäts hjärtats elektriska aktivitet och visas som vågor med benämningen ”P”, ”Q”, ”R”, ”S” och ”T”.QT-intervallet är ett mått mellan Q-vågens början och T-vågens slut.
Studiens utformning
TQT-studier utförs in vivo för alla nya molekylära enheter (NME).De måste utföras före fas III-prövningar, oberoende av in vitro- och icke-kliniska fynd.
I regel utförs TQT-studier som crossover-studier med engångsdos hos friska deltagare.Forskare utvärderar terapeutiska och supraterapeutiska (mer än terapeutisk) doser av läkemedlet jämfört med en positiv kontroll (till exempel ett vanligt antibiotikum som moxifloxacin) och en negativ kontroll (en placebo).
A2-5.03.4-V1.1