Last update: 29 September 2016


Einleitung
Die während der frühen klinischen Entwicklung durchgeführten Studien (Phase I und Phase II) haben eine Vielzahl von unterschiedlichen Zielsetzungen. Diese frühen klinischen Studien müssen vor allem zeigen, dass ein Prüfpräparat für den Menschen sicher ist. In diesen Studien wird auch versucht, zu zeigen, dass das Arzneimittel gegen die beabsichtigte Erkrankung oder den Zustand wirksam ist.
Folgende Schlüsselfragen müssen während der frühen klinischen Entwicklung beantwortet werden:
- Phase I
- Ist das Arzneimittel sicher beim Menschen? Auf welchen Stufen? (Toleranz)
- Was macht der Körper mit dem Arzneimittel? (Pharmakokinetik [PK])
- Was macht das Arzneimittel mit dem Körper? (Pharmakodynamik [PD])
- Welche Wechselwirkungen gibt es? (Wirkstoff-Wirkstoff-Wechselwirkungen, Wechselwirkungen mit Lebensmitteln, usw.)
- Ist das Arzneimittel aktiv?
- Phase II
- Ist das Arzneimittel sicher bei Patienten? (Sicherheit)
- Was macht das Arzneimittel mit dem Körper? (Pharmakodynamik [PD])
- Scheint das Arzneimittel bei Patienten zu wirken? Bei welchen Dosen? (Wirkung)
- Wie sollten konfirmatorische Prüfungen gestaltet werden? (Endpunkte, Zielpopulation, andere Arzneimittel, die eingenommen werden [begleitend], usw.)
Obwohl die Arzneimittelentwicklung typischerweise als eine chronologische Folge von Phasen dargestellt wird, werden die Studien, die in den jeweiligen Phasen durchgeführt werden, normalerweise anhand ihrer Zielsetzungen klassifiziert. Wie im nachstehenden Diagramm dargestellt, können Studien, die normalerweise Teil einer früheren Phase sind, später im Prozess der Arzneimittelentwicklung durchgeführt werden, wenn neue Daten auf die Notwendigkeit für zusätzliche Informationen hindeuten.
Eskalationsstudien mit ansteigender Einzeldosis/Mehrfachdosis (SAD und MAD)
Einzeldosis-Eskalationsstudien (SAD) und Mehrfachdosis-Eskalationsstudien (MAD) sind normalerweise die ersten, am Menschen durchgeführten Studien.
Studienziele
SAD und MAD-Studien:
- Untersuchung von Sicherheit und Verträglichkeit
- Identifizierung einer maximal verträglichen Dosis (MTD – Maximum Tolerated Dose)
- Untersuchung der allgemeinen pharmakokinetischen (PK) Merkmale
- Zu untersuchen, wie ein stabiler Spiegel des Arzneimittels im Körper im Laufe der Zeit erreicht werden kann. Diese Bedingungen sind als Steady-State-Parameter (Akkumulationszeit-Abhängigkeit) bekannt.
- Durchführung einer vorläufigen Untersuchung der Arzneimittelausscheidung aus dem Körper (Identifizierung von Metaboliten)
Studiendesign
Die Anfangsdosis für SAD/MAD-Studien wird anhand der Ergebnisse der nicht-klinischen toxikologischen Studien bestimmt. Die Dosis wird dann gemäß den Eskalationsschemata erhöht, die in den regulatorischen Guidance-Dokumenten festgelegt sind. Die Studien werden gemäß Abbruchregeln abgebrochen, die Toxizität und das Fehlen von Toxizität (maximierte Exposition, maximierte Pharmakodynamik, usw.) umfassen.
'Massenbilanz'-Studien: Absorption, Distribution, Metabolismus und Ausscheidung (ADME – Absorption, Distribution, Metabolism, Excretion)
Studienziele
ADME-Studien werden mit dem Ziel durchgeführt, das pharmakokinetische (PK) Profil des Prüfpräparates zu verstehen und zu charakterisieren: das heißt, was der Körper mit dem Arzneimittel macht. Diese Studien untersuchen die Art und Weise, wie der Körper das Arzneimittel absorbiert, wie das Arzneimittel im ganzen Körper verteilt wird, wie der Körper das Arzneimittel verstoffwechselt, und wie der Körper das Arzneimittel ausscheidet (siehe Bild unten).
-
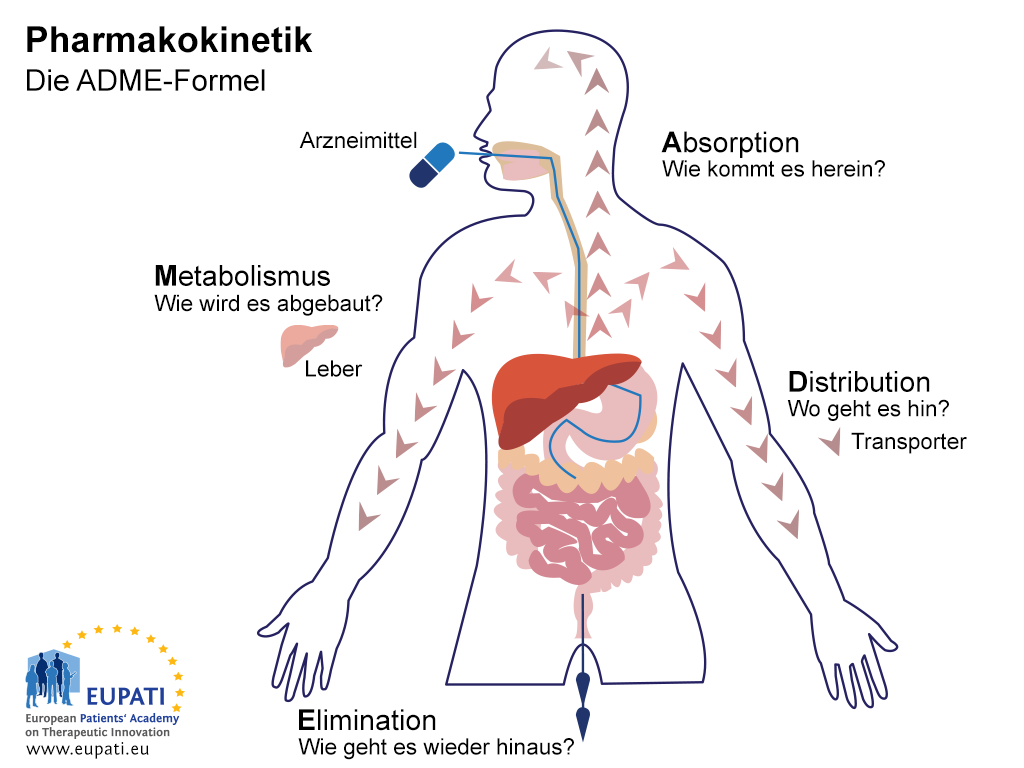
- Die Grundsätze der Pharmakokinetik – also der Lehre davon, wie der Körper mit einem Arzneimittel umgeht – spiegeln sich in dem Kürzel ADME wider.
Studiendesign
ADME-Studien werden in der Regel mit einer Einzeldosis des Arzneimittels in einer kleinen Gruppe (normalerweise vier bis sechs Probanden) von gesunden Männern (um mögliche Schäden für Frauen im gebärfähigen Alter auszuschließen) über den beabsichtigten Verabreichungsweg durchgeführt. Diese Studien werden in der Regel mit Phase I der Entwicklung assoziiert, können jedoch zu jeder Zeit während der Entwicklung eines Arzneimittels durchgeführt werden.
ADME-Studien liefern auch Informationen über die Bioverfügbarkeit – das heißt, den Anteil der verabreichten Dosis des aktiven Bestandteils des Arzneimittels, der in die Blutbahn gelangt.
Bioverfügbarkeits- und Bioäquivalenz-Studien
Studienziele
Bioverfügbarkeitsstudien bewerten die Geschwindigkeit und das Ausmaß der Resorption eines Arzneimittels. Sie untersuchen die Konzentration des aktiven pharmazeutischen Bestandteils in der Blutbahn im Verlauf der Zeit.
Arzneimittel, die über verschiedene Wege verabreicht werden, haben unterschiedliche Bioverfügbarkeitsprofile. So weisen beispielsweise Arzneimittel, die direkt in die Blutbahn, z. B. per intravenöser Injektion (i.v.) verabreicht werden, eine Bioverfügbarkeit von 100 % auf, sobald sie verabreicht werden, wohingegen Arzneimittel, die oral verabreicht werden, nicht bioverfügbar sind, bis das Arzneimittel nach Resorption in die Blutbahn gelangt.
-
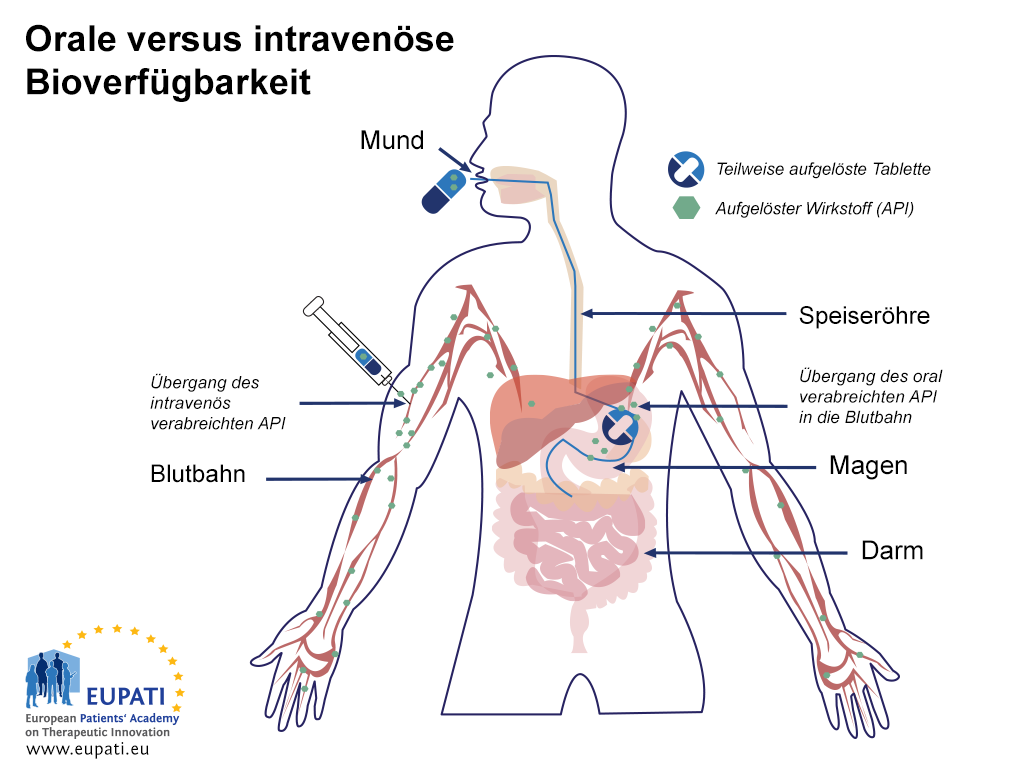
- Eine schematische Darstellung, welche die Resorption einer oral verabreichten Kapsel im Vergleich zu einer direkt in die Blutbahn verabreichten Injektion (intravenöse Injektion) zeigt. Nach Erreichen des Magens wird die Kapsel in den Dünndarm weitertransportiert, wo die weitere Resorption stattfindet.
Bioverfügbarkeitsstudien bewerten die Geschwindigkeit der Resorption eines Arzneimittels, indem die höchste Konzentration des Arzneimittels (Cmax) in der Blutbahn und die Zeit, zu der diese höchste Konzentration vorliegt (Tmax), gemessen wird. Die Fläche unter der Kurve (AUC) stellt die Gesamtexposition des Körpers gegenüber dem Arzneimittel dar und wird verwendet, um das Ausmaß der Resorption zu untersuchen.
-
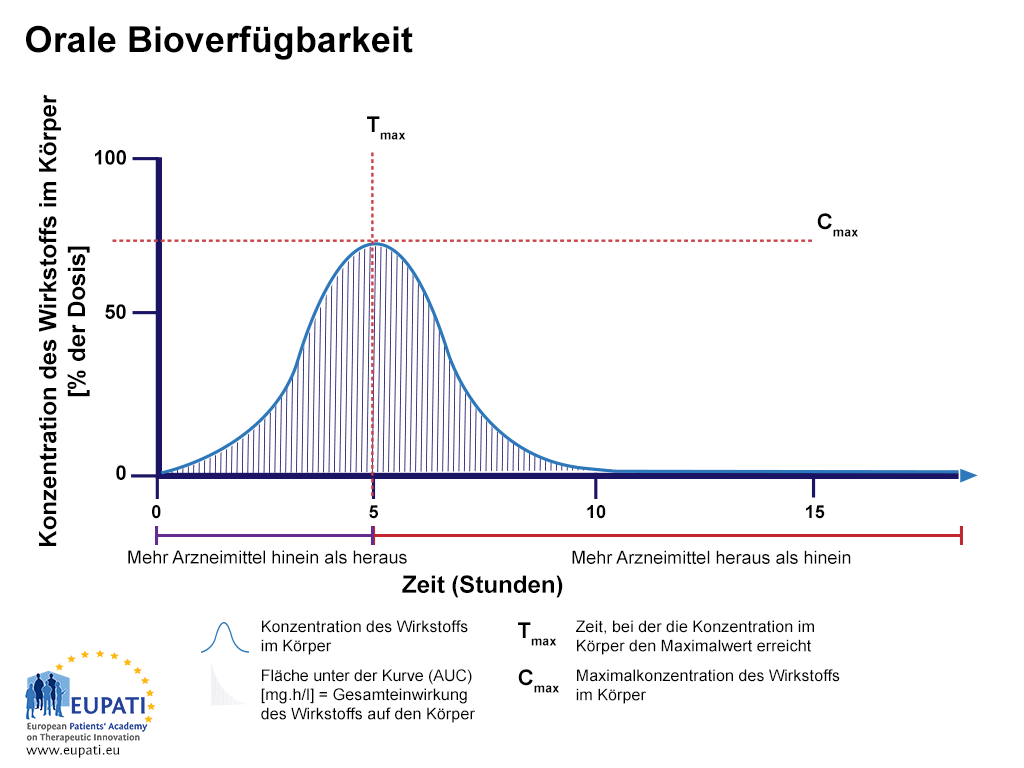
- Der Prozentsatz des Wirkstoffs nach Schlucken einer Tablette, untersucht über einen Zeitraum von 15 Stunden. Die Fläche unter der Kurve (AUC) ist schattiert dargestellt. Tmax bezeichnet den Zeitpunkt, zu dem die höchste Konzentration des Wirkstoffs im Blut gemessen wird, wohingegen Cmax die maximale Konzentration des Wirkstoffs in der Blutbahn bezeichnet.
Studiendesign
Diese Studien werden normalerweise als Crossover-, randomisierte, Einzeldosis-Studien an gesunden Teilnehmern durchgeführt. Die Forscher messen die Konzentration des Arzneimittels und seine wesentlichen aktiven Metaboliten im Blut und Plasma der Teilnehmer.
Bioäquivalenzstudien
Bioäquivalenzstudien untersuchen die Beziehung zwischen zwei verschiedenen Formulierungen. Sie untersuchen auch die Geschwindigkeit und das Ausmaß der Resorption eines Arzneimittels, aber sie vergleichen diese mit der Geschwindigkeit und dem Ausmaß der Resorption eines anderen Arzneimittels oder einer anderen Formulierung des gleichen Arzneimittels (Referenzformulierung/Arzneimittel). Bioäquivalenzstudien werden verwendet, um Generika mit ihren Referenzarzneimitteln zu vergleichen. Es gibt festgelegte Kriterien, die ein Arzneimittel erfüllen muss, bevor es mit einem anderen Arzneimittel als bioäquivalent angesehen werden kann.
Studien zu Lebensmittelwirkungen
Studienziele
Studien zu Lebensmittelwirkungen bewerten die Wirkung von Lebensmitteln auf die Geschwindigkeit, das Ausmaß und die Bioverfügbarkeit der Resorption eines Arzneimittels in einer bestimmten Formulierung. Die aus Studien zu Lebensmittelwirkungen gewonnenen Informationen sind wichtig für die Anweisungen zur Verabreichung, die in der Packungsbeilage enthalten sind, ob das Arzneimittel auf nüchternen Magen oder mit einer Mahlzeit eingenommen werden sollte.
Studiendesign
Diese Studien sind normalerweise Crossover-Einzeldosis-Studien, die zwei verschiedene Bedingungen vergleichen: Teilnehmer mit nüchternem Magen mit Teilnehmern, die eine Mahlzeit mit hohem Fett- und Kaloriengehalt eingenommen haben. Die Studie hat in der Regel zwei Sequenzen und wird an gesunden Teilnehmern mit der höchsten erwarteten Stärke des Arzneimittels durchgeführt.
Studien zu Nierenfunktionsbeeinträchtigung
Studienziele
Studien zu Nierenfunktionsbeeinträchtigung werden mit dem Ziel, das Arzneimittel in Menschen mit unterschiedlichen Stufen der Nierenfunktion zu bewerten, durchgeführt. Diese Studien sammeln Informationen über die Auswirkungen einer Nierenfunktionsbeeinträchtigung auf die Ausscheidung von Arzneimitteln aus dem Körper und Dosierungsempfehlungen für Patienten mit verschiedenen Stadien der Nierenfunktionsbeeinträchtigung.
Studiendesign
Studien zu Nierenfunktionsbeeinträchtigung werden als Einzeldosis-Parallelgruppenstudien an freiwilligen, gesunden männlichen und weiblichen Probanden (ungefähr sechs pro Gruppe) durchgeführt. Die Gruppen werden anhand der Biomarker für die Nierenfunktion stratifiziert.
Studien zur Beeinträchtigung der Leber
Studienziele
Studien zur Beeinträchtigung der Leber werden mit dem Ziel, das Arzneimittel in Menschen mit unterschiedlichen Stufen der Beeinträchtigung der Leber zu bewerten, durchgeführt. Diese Studien untersuchen die Wirkung der Beeinträchtigung der Leber auf die Pharmakokinetik des Arzneimittels und dessen Metaboliten und liefern Dosierungsempfehlungen für verschiedene Stadien der Beeinträchtigung der Leber aus Gründen der Wirksamkeit und/oder der Sicherheit.
Studiendesign
Sind die pharmakokinetischen Ergebnisse früherer Studien linear und zeitunabhängig, werden die Studien zur Beeinträchtigung der Leber typischerweise als Parallelgruppen-Studien in gesunden männlichen und weiblichen Probanden (etwa acht Personen) mit unterschiedlichem Grad der Beeinträchtigung der Leberfunktion durchgeführt. Die Behandlungsgruppen werden anhand der Standardklassifikationen von Leberinsuffizienz stratifiziert.
Wird das Arzneimittel durch ein Enzym metabolisiert, das nur aufgrund der genetischen Variation auftritt, dann sollten die Teilnehmer auf der Grundlage ihres Genotyp-Status beurteilt werden.
Studien zu Arzneimittelwechselwirkungen (Wirkstoff-Wirkstoff-Wechselwirkungen)
Studienziele
Studien zu Arzneimittelwechselwirkungen (gemeinhin bekannt als Wirkstoff-Wirkstoff-Wechselwirkungsstudien) untersuchen die Wirkung einer Begleitmedikation auf die Pharmakokinetik des Prüfpräparates (IMP) sowie die Wirkung des IMP auf die Pharmakokinetik der Begleitmedikation.
Studiendesign
Diese Studien basieren auf früheren in-vitro-Ergebnissen und werden vorzugsweise mit einem Crossover-Studiendesign durchgeführt. Studien zu Arzneimittelwechselwirkungen werden in gesunden, freiwilligen Probanden oder in Patienten durchgeführt, wenn die Bewertung der pharmakodynamischen Endpunkte erwünscht ist, sollten die Arzneimittel zu toxisch sein (zum Beispiel Anti-Krebs-Arzneimittel).
Diese Studien haben normalerweise ein Crossover-Design. Dosierung, Dosierungsintervalle, Anzahl der Dosen, Verabreichungswege und Zeitpunkt der gleichzeitigen Verabreichung sollten so gewählt werden, dass die Möglichkeiten, eine Wechselwirkung festzustellen, maximiert und die klinische Umgebung nachgeahmt werden. Das Ausmaß der Wechselwirkung (Hemmung/Einleitung) wird durch die Änderung der Resorption eines der Prüfpräparate eingestuft, berechnet als die Fläche unter der Kurve (AUC).
Thorough QT-Studie (TQT)
Studienziele
Ein QT-Intervall ist ein Maß des Herzrhythmus. Ein QT-Intervall kann mithilfe eines Elektrokardiogramms (EKG) gemessen und als (unvollkommener) Biomarker verwendet werden, um das Risiko eines Arzneimittels, Arrhythmie auszulösen, zu bewerten. In einem EKG wird die elektrische Aktivität des Herzens gemessen und in Form von Wellen, bezeichnet als „P”, „Q”, „R”, „S” sowie „T” dargestellt. Das QT-Intervall ist eine Messung zwischen dem Beginn der Q-Welle und dem Ende der T-Welle.
Studiendesign
TQT-Studien werden in-vivo für sämtliche neuen Wirkstoffe durchgeführt (NMEs, New Molecular Entities). Sie müssen vor Phase-III-Studien, unabhängig von den in-vitro- bzw. nicht-klinischen Erkenntnissen stattfinden.
TQT-Studien werden normalerweise als Crossover-Einzeldosis-Studien an gesunden Teilnehmern durchgeführt. Forscher bewerten therapeutische und supratherapeutisch (größer als die therapeutischen) Dosen des Arzneimittels gegenüber einer Positivkontrolle (beispielsweise ein gewöhnliches Antibiotikum wie Moxifloxacin) und einer Negativkontrolle (Placebo).
A2-5.03.4-V1.1