Last update: 29 septiembre 2016


Introducción
Los estudios realizados normalmente durante el desarrollo clínico inicial (fase I y fase II) tienen una amplia variedad de objetivos. Estos ensayos clínicos iniciales deben establecer ante todo que el medicamento en investigación es seguro para los seres humanos. Además, tratan de demostrar que el medicamento es eficaz para combatir la enfermedad o el trastorno correspondientes.
Durante el desarrollo clínico inicial se debe responder a las siguientes preguntas clave:
- Fase I
- ¿El medicamento es seguro en seres humanos? ¿En qué medida? (Tolerancia)
- ¿Cómo procesa el organismo el fármaco? (Farmacocinética [FC])
- ¿Cuál es el mecanismo de acción del fármaco en el organismo? (Farmacodinámica [FD])
- ¿Qué interacciones se producen? (Interacciones farmacológicas, interacciones con alimentos y bebidas, etc.)
- ¿El medicamento es activo?
- Fase II
- ¿El medicamento es seguro en pacientes? (Seguridad)
- ¿Cuál es el mecanismo de acción del fármaco en el organismo? (Farmacodinámica [FD])
- ¿Parece funcionar el medicamento en pacientes? ¿Con qué dosis? (Efecto)
- ¿Cómo se deben diseñar los ensayos de confirmación? (Criterios de valoración, población de destino, otros medicamentos administrados (simultáneamente), etc.)
Aunque el desarrollo de fármacos se suele representar como una serie cronológica de fases, los estudios realizados en cada fase se suelen clasificar según sus objetivos. Como se muestra en el diagrama siguiente, los estudios que suelen formar parte de una fase inicial se pueden realizar posteriormente durante el proceso de desarrollo de fármacos a medida que los nuevos datos indiquen la necesidad de información adicional.
Estudios de dosis única ascendente y dosis múltiples ascendentes (DUA y DMA)
Los estudios de dosis única ascendente y dosis múltiples ascendentes son normalmente estudios de primer ensayo clínico en el ser humano.
Objetivos de los estudios
Estudios de DUA y DMA:
- Investigar la seguridad y la tolerabilidad.
- Identificar la dosis máxima tolerada (DMT).
- Investigar las características farmacocinéticas (FC) generales.
- Investigar cómo se puede alcanzar un nivel estable del fármaco en el organismo con el tiempo. Estas condiciones se conocen como parámetros de situación de equilibrio (acumulación dependiente del tiempo).
- Realizar una exploración preliminar de la eliminación del fármaco del organismo (identificación de metabolitos).
Diseño de los estudios
La dosis inicial para los estudios de dosis única ascendente y dosis múltiples ascendentes se determina en función de los resultados de los estudio de toxicología no clínicos. A continuación, la dosis se aumenta según los patrones de aumento establecidos en los documentos de directrices regulatorias. Los estudios se paran según las reglas de parada, lo que incluye la toxicidad y la ausencia de toxicidad (exposición maximizada, farmacodinámica maximizada, etc.).
Estudios de «balance de masas»: absorción, distribución, metabolismo y eliminación (ADME)
Objetivos de los estudios
El objetivo de los estudios de ADME es comprender y caracterizar el perfil farmacocinético (FC) del medicamento en investigación (es decir, cómo procesa el organismo el fármaco). En estos estudios se investiga el modo en el que el organismo absorbe el fármaco, cómo se distribuye el fármaco en el organismo, cómo metaboliza el organismo el fármaco y cómo elimina el organismo el fármaco (consulte la imagen siguiente).
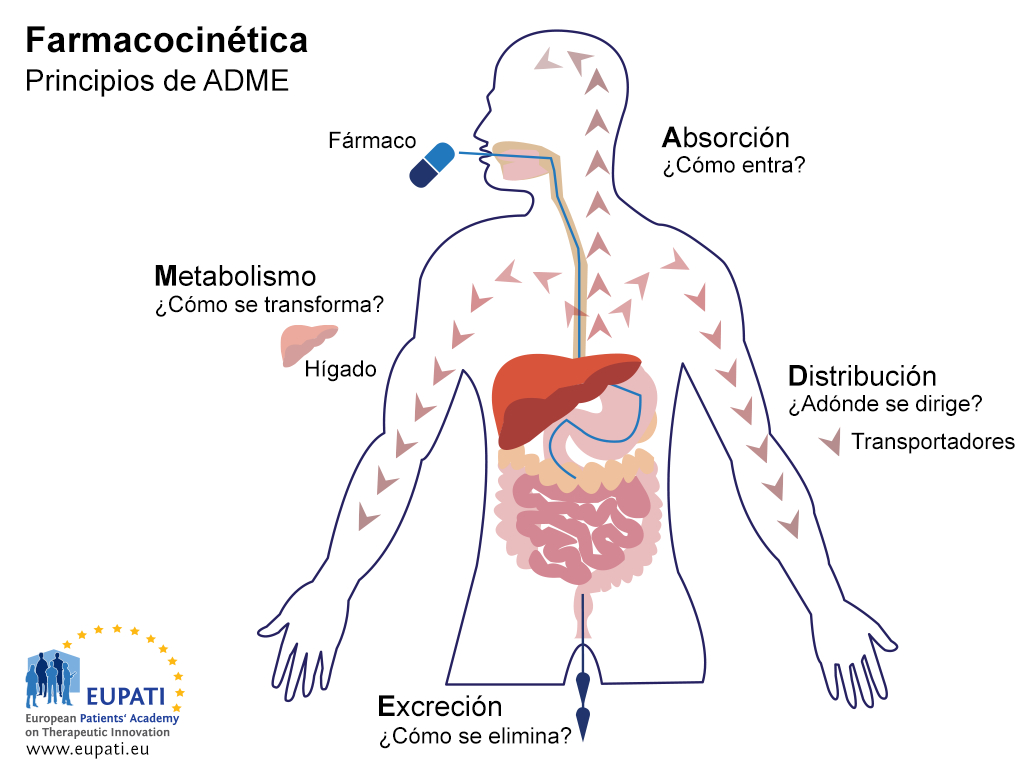
Los principios clave de la Farmacocinética - el estudio del efecto que el cuerpo tiene sobre un medicamento - están representados en el acrónimo ADME.
Diseño de los estudios
Los estudios de ADME se suelen realizar con una dosis única del fármaco administrada a un número reducido de hombres sanos (normalmente, de cuatro a seis) (para excluir la posibilidad de efectos perjudiciales en las mujeres en edad fértil) mediante la vía de administración prevista. Estos estudios se suelen asociar a la fase I de desarrollo, pero se pueden realizar durante todo el desarrollo de un medicamento.
Además, los estudios de ADME proporcionan información sobre la biodisponibilidad (es decir, la fracción de una dosis administrada del principio activo que llega al torrente circulatorio).
Estudios de biodisponibilidad (BD) y bioequivalencia (BE)
Objetivos de los estudios
En los estudios de biodisponibilidad se evalúan la velocidad y el nivel de absorción de un fármaco. Se investiga, además, la concentración del principio activo en el torrente circulatorio con el tiempo.
Los fármacos administrados mediante distintas vías tienen perfiles de biodisponibilidad diferentes. Por ejemplo, los fármacos administrados directamente en el torrente circulatorio mediante una inyección intravenosa (IV) tienen una biodisponibilidad del 100% en cuanto se administran, mientras que los fármacos administrados por vía oral no están biodisponibles hasta que el fármaco llega al torrente circulatorio después de ser absorbido.
En los estudios de biodisponibilidad se evalúa la velocidad de absorción de un fármaco mediante la medición de la concentración máxima del fármaco (Cmáx) en el torrente circulatorio y el tiempo hasta la concentración máxima (Tmáx). El área bajo la curva (ABC) representa la exposición total del organismo al fármaco y se usa para analizar el grado de absorción.
Diseño de los estudios
Estos estudios se suelen realizar como estudios de dosis única aleatorizados con grupos cruzados (participantes sanos). Los investigadores miden la concentración del fármaco y sus metabolitos activos principales en la sangre y el plasma de los participantes.
Estudios de bioequivalencia
En los estudios de bioequivalencia se investiga la relación entre dos formulaciones diferentes. Además, se estudian la velocidad y el nivel de absorción de un fármaco, pero se comparan con la velocidad y el nivel de absorción de otro fármaco o una formulación diferente del mismo fármaco (formulación/fármaco de referencia). Los estudios de bioequivalencia se usan para comparar los medicamentos genéricos con los medicamentos de referencia. Hay una serie de criterios establecidos que un fármaco debe cumplir para poder establecer una relación de bioequivalencia con otro fármaco.
Estudios del efecto de los alimentos
Objetivos de los estudios
En estos estudios se evalúa el efecto de los alimentos en la velocidad, el nivel y la biodisponibilidad de la absorción del fármaco con una formulación determinada. La información de los estudios del efecto de los alimentos es importante para las instrucciones de administración del prospecto, donde se indica si el medicamento se debe tomar en ayunas o con las comidas.
Diseño de los estudios
Estos estudios se suelen realizar con una dosis única y grupos cruzados para comparar dos condiciones (participantes en ayunas en comparación con participantes que han ingerido alimentos con un alto contenido de grasas y un alto aporte calórico). El estudio suele constar de dos secuencias y se realiza con participantes sanos y la máxima concentración prevista del fármaco.
Estudios de insuficiencia renal
Objetivos de los estudios
El objetivo de los estudios de insuficiencia renal es evaluar el fármaco en personas con niveles variables de la función renal (riñones). En estos estudios se recopila información sobre el efecto de la insuficiencia renal en la eliminación del fármaco del organismo y las recomendaciones de dosis para pacientes con distintos grados de insuficiencia renal.
Diseño de los estudios
Los estudios de insuficiencia renal son estudios de dosis única y grupos paralelos realizados con voluntarios sanos (hombres y mujeres) (unos seis por grupo). Los grupos se estratifican según los biomarcadores de la función renal.
Estudios de insuficiencia hepática
Objetivos de los estudios
El objetivo de los estudios de insuficiencia hepática es evaluar el fármaco en personas con niveles variables de insuficiencia hepática (hígado). En estos estudios se investiga el efecto de la insuficiencia hepática en la farmacocinética del fármaco y sus metabolitos, y se establecen recomendaciones de dosis para los distintos grados de insuficiencia hepática por motivos de eficacia y seguridad.
Diseño de los estudios
Si los resultados de farmacocinética de estudios anteriores son lineales e independientes del tiempo, los estudios de insuficiencia hepática se suelen realizar como estudios de grupos paralelos con voluntarios sanos (hombres y mujeres) (unos ocho voluntarios) con distintos grados de insuficiencia hepática. Los grupos de tratamiento se estratifican según la clasificación de referencia de la insuficiencia hepática.
Si el fármaco es metabolizado por una enzima que solo se da por una variación genética, los participantes se deben evaluar según su genotipo.
Estudios de interacciones con otros medicamentos (interacción farmacológica, IF)
Objetivos de los estudios
Los estudios de interacciones con otros medicamentos (normalmente llamados estudios de interacción farmacológica) permiten evaluar el efecto de la administración de varios fármacos en la farmacocinética del medicamento en investigación y el efecto del medicamento en investigación en la farmacocinética de los demás fármacos.
Diseño de los estudios
Estos estudios se basan en resultados in vitro anteriores y se realizan preferiblemente con un diseño de grupos cruzados. Los estudios de interacción farmacológica se realizan con voluntarios sanos o pacientes si se recomienda evaluar los criterios de valoración de la farmacodinámica cuando los fármacos son demasiado tóxicos (por ejemplo, fármacos contra el cáncer).
Normalmente, estos estudios tienen un diseño de grupos cruzados. La dosis, los intervalos de administración, el número de dosis, las vías de administración y la duración del tratamiento simultáneo con varios fármacos se deben determinar para maximizar la posibilidad de detectar una interacción y deben reproducir las condiciones del entorno clínico. El grado de interacción (inhibición/inducción) se clasifica según la variación de la absorción de uno de los medicamentos en investigación, calculada como el área bajo la curva (ABC) de la sustancia.
Estudios de QT completos (QTC)
Objetivos de los estudios
Un intervalo QT es una medición del ritmo cardíaco. Un intervalo QT se puede medir mediante un electrocardiograma (ECG) y se puede usar como un biomarcador (imperfecto) para evaluar el riesgo de que un fármaco provoque arritmia. En un ECG, la actividad eléctrica del corazón se mide y muestra en forma de ondas etiquetadas como P, Q, R, S y T. El intervalo QT es una medición entre el inicio de la onda Q y el final de la onda T.
Diseño de los estudios
Los estudios QTC se realizan in vivo para entidades moleculares nuevas. Se deben realizar antes de los ensayos de fase III con independencia de los resultados in vitro o no clínicos.
En general, los estudios de QTC se realizan con una dosis única y grupos cruzados con participantes sanos. Los investigadores evalúan las dosis terapéuticas y supraterapéuticas (superiores a las terapéuticas) del fármaco en comparación con una referencia positiva (por ejemplo, un antibiótico común como la moxifloxacina) y una referencia negativa (placebo).
A2-5.03.4-V1.1