

Introduction
The studies typically conducted during early clinical development (Phase I and Phase II) have a variety of objectives. These early clinical trials must establish above all that an investigational medicinal product is safe for humans. They also try to show that the medicine is effective against the intended disease or condition.
The following key questions must be answered during early clinical development:
- Phase I
- Is the medicine safe in humans? At what levels? (Tolerance)
- What does the body do to the medicine? (Pharmacokinetics (PK))
- What does the medicine do to the body? (Pharmacodynamics (PD))
- What interactions are there? (Drug-Drug interactions, interactions with food and drink, etc.)
- Is the medicine active?
- Phase II
- Is the medicine safe in patients? (Safety)
- What does the medicine do to the body? (Pharmacodynamics (PD))
- Does the medicine seem to work in patients? At what dose(s)? (Effect)
- How should confirmatory trials be designed? (Endpoints, target population, other medications being taken (concomitant), etc.)
Although medicines development is typically represented as a chronological series of phases, the studies that are performed within each phase are typically classified based on their objectives. As shown in the diagram below, studies that are typically part of an earlier phase may be performed later on in the medicines development process as emerging data suggests the need for additional information.
-
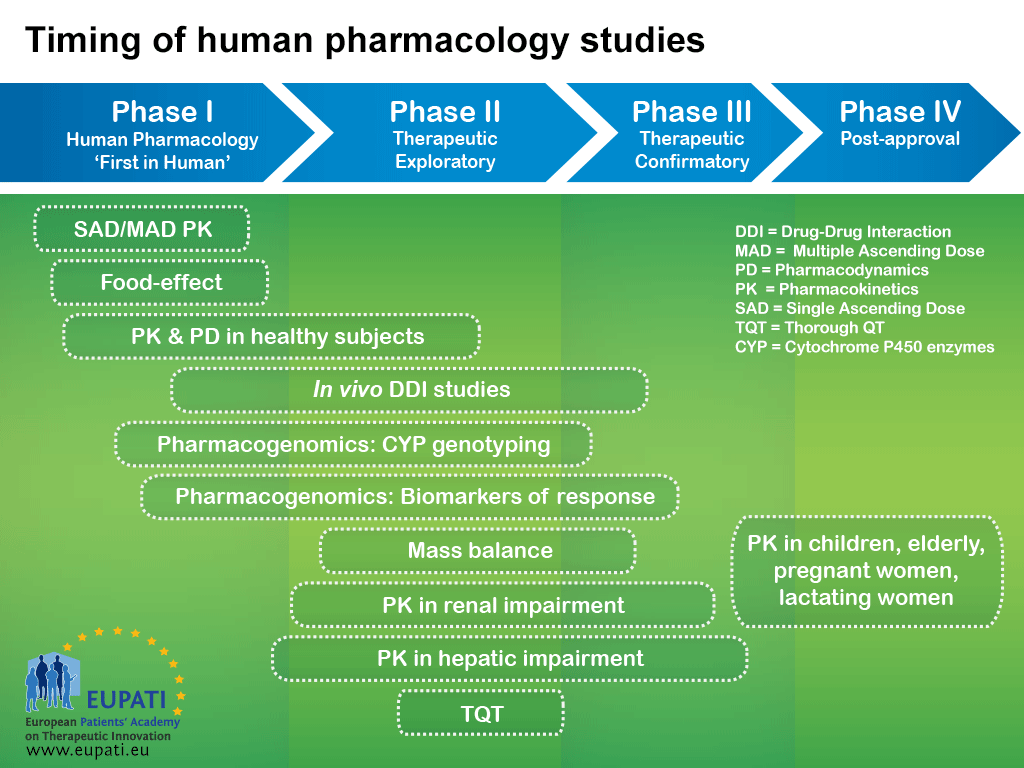
- The logic behind representing medicines development in a series of consecutive phases comes from the idea that the results of prior studies should influence the plans for later studies: emerging data will frequently initiate modifications of the development strategies.
Single Dose/Multiple Ascending Dose escalation studies (SAD and MAD)
Single Ascending Dose (SAD) and Multiple Ascending Dose (MAD) studies are usually the first-in-human studies.
Study objectives
- Investigate safety and tolerability
- Identify a Maximum Tolerated Dose (MTD)
- Investigate general pharmacokinetic (PK) characteristics
- Investigate how to achieve a stable level of medicine in the body over time, these conditions are known as steady-state parameters (accumulation time-dependency)
- Perform preliminary exploration of medicine excretion from the body (identifying metabolite(s))
Study design
The starting dose for SAD/MAD studies is determined based on the results of non-clinical toxicology studies. The dose is then increased according to escalation schemes which are laid out in regulatory guidance documents. The studies are stopped according to stopping rules, which include toxicity and absent toxicity (maximised exposure, maximised pharmacodynamics, etc.).
‘Mass balance’ studies: Absorption, Distribution, Metabolism, Excretion (ADME)
Study objectives
The objective of ADME studies is to understand and characterise the pharmacokinetic (PK) profile of the investigational medicinal product: that is, what the body does to the medicine. These studies investigate the way the body absorbs the medicine, how the medicine is distributed throughout the body, how the body metabolises the medicine, and how the body excretes the medicine (see image below).
-
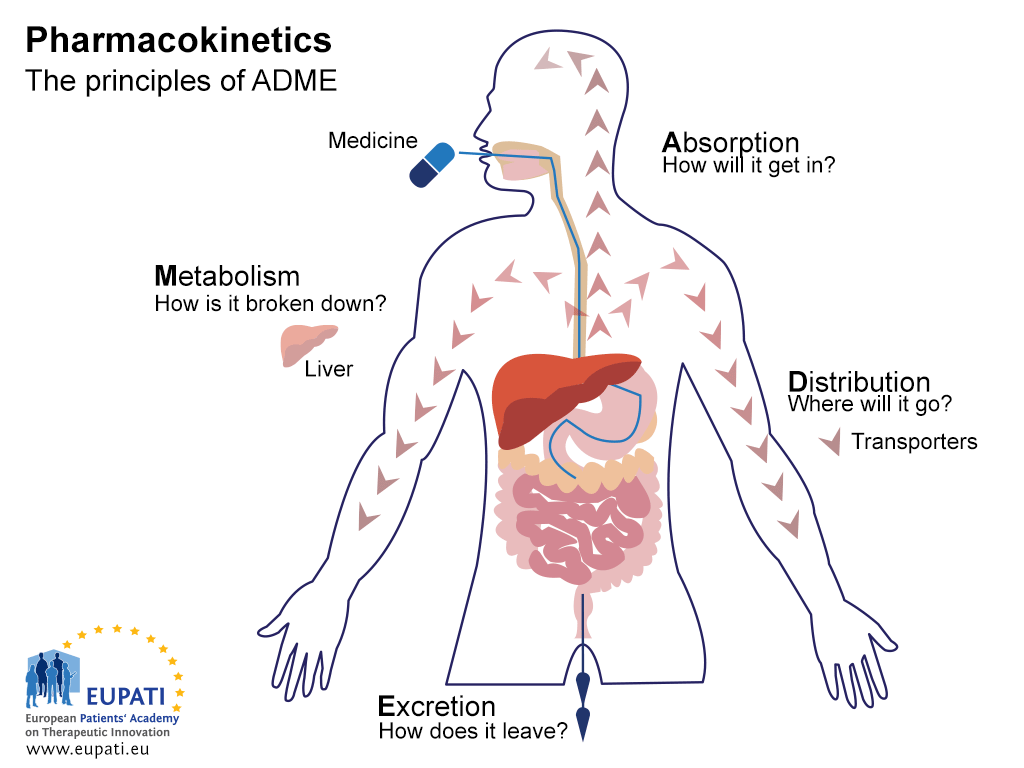
- The key principles of Pharmacokinetics – the study of the effect the body has on a medicine – are represented in the acronym ADME.
Study design
ADME studies are usually performed using a single dose of the medicine in a small number (usually four to six) of healthy males (to exclude possible harm to women of child bearing potential), through the intended route of administration. These studies are typically associated with Phase I of development, but they may be performed throughout the development of a medicinal product.
ADME studies also provide information on bioavailability – that is, the fraction of an administered dose of active pharmaceutical ingredient (API) that reaches the bloodstream.
Bioavailability (BA) and Bioequivalence (BE) studies
Study objectives
Bioavailability studies evaluate the rate and extent of absorption of a medicine. They investigate the concentration of the active pharmaceutical ingredient (API) in the bloodstream over time.
Medicines administered via different routes have different bioavailability profiles. For instance, medicines that are administered directly into the bloodstream through intravenous injection (IV) have a 100% bioavailability as soon as they are administered, whereas medicines that are administered orally are not bioavailable until the medicine enters the bloodstream after it is absorbed.
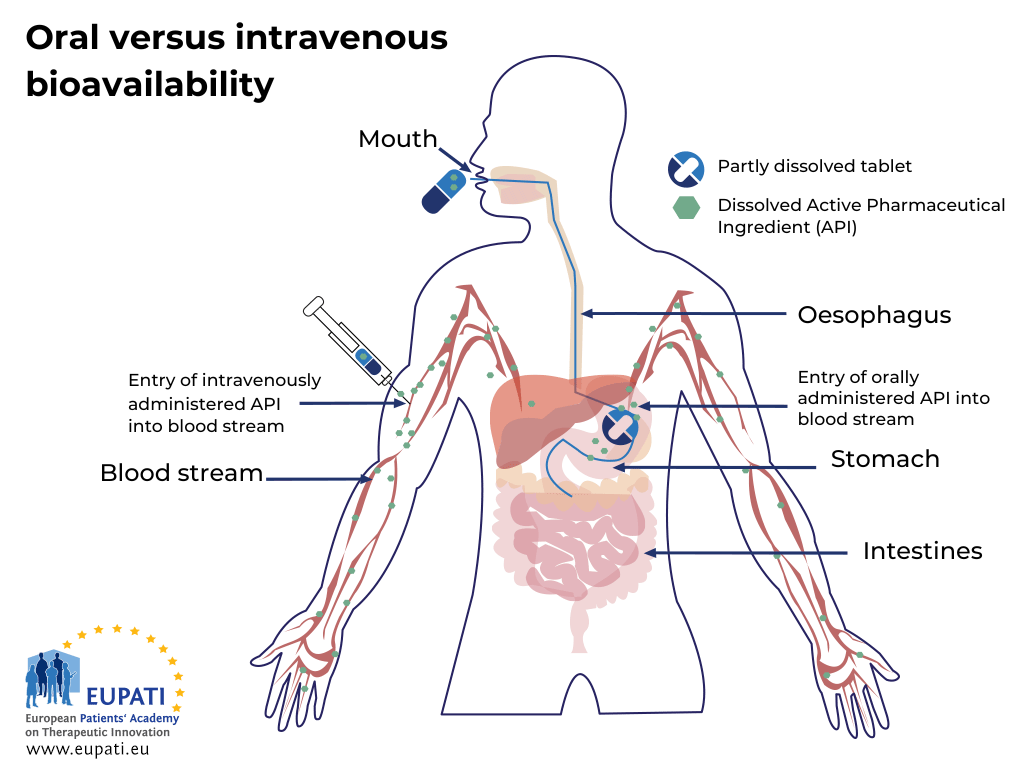
Comparing the bioavailability of Active Pharmaceutical Ingredients administered orally and intravenously. An API is said to be ‘biologically available’ (bioavailable) when it enters the blood stream.
Bioavailability studies assess the rate of absorption of a medicine by measuring the maximum concentration of the medicine (Cmax) in the bloodstream and the time at which the maximum concentration occurs (Tmax). The area under the curve (AUC) represents the total exposure of the body to the medicine and is used to study the extent of absorption.
Study design
These studies are typically run as crossover, randomised, single-dose studies in healthy participants. Researchers measure the concentration of the medicine and its major active metabolites in participants’ blood and plasma.
Bioequivalence studies
Bioequivalence studies investigate the relationship between two different formulations. They also study the rate and extent of absorption of a medicine, but they compare these with the rate and extent of absorption of another medicine or a different formulation of the same medicine (reference formulation/medicine). Bioequivalence studies are used to compare generic medicines with their reference medicines. There are set criteria that a medicine must fulfil before it can be considered bioequivalent with another medicine.
Food effect studies
Study objectives
Food effect studies evaluate the effect of food on the rate, extent, and bioavailability of medicine absorption in a given formulation. The information from food effect studies is important for the administration instructions that are put in the package leaflet (PL) instructing whether the medicine should be taken on an empty stomach or with meals.
Study design
These studies are typically single dose, crossover studies that compare two conditions: participants who have fasted versus participants fed with a high-fat, high-calorie meal. The study usually has two sequences, and is performed in healthy participants using the highest anticipated strength of the medicine.
Renal impairment studies
Study objectives
The objective of renal impairment studies is to evaluate the medicine in people with varying levels of renal (kidney) function. These studies gather information on the effect of renal impairment on the excretion of medicine from the body and dosage recommendations for patients with various stages of renal impairment.
Study design
Renal impairment studies are run as single dose, parallel group studies in healthy male and female volunteers (around six per group). The groups are stratified based on renal function biomarkers.
Hepatic impairment studies
Study objectives
The objective of hepatic impairment studies is to evaluate the medicine in people with different levels of hepatic (liver) impairment. These studies examine the effect of hepatic impairment on the pharmacokinetics of the medicine and its metabolites, and contribute dosage recommendations for various stages of hepatic impairment for efficacy and/or safety reasons.
Study design
If the pharmacokinetic results of previous studies are linear and time-independent, then hepatic impairment studies are typically conducted as parallel group studies with healthy male and female volunteers (around eight individuals) with varying degrees of impairment of hepatic function. The treatment groups are stratified based on standard classifications of hepatic impairment.
If the medicine is metabolised by an enzyme that only occurs due to genetic variation, then participants should be assessed based on their genotype status.
Medicines interaction (Drug-Drug Interaction, DDI) studies
Study objectives
Medicines interaction studies (commonly known as Drug-Drug Interaction (DDI) studies) evaluate the effect of concomitant medication on the pharmacokinetics of the investigational medicinal product (IMP) as well as the effect of the IMP on the pharmacokinetics of concomitant medications.
Study design
These studies are guided by previous in vitro results; they are preferably conducted using a crossover trial design. Medicines interaction studies are conducted in healthy volunteers or in patients if it is desirable to evaluate pharmacodynamic endpoints when medicines are too toxic (for instance, anti-cancer medicine).
Typically, these studies use a crossover trial design. The dosage, dosing intervals, number of doses, routes, and timing of co-administration should be designed to maximise the possibility of detecting an interaction and should mimic the clinical setting. The amount of interaction (inhibition/induction) is classified by the change in absorption of one of the investigational medicinal products, calculated as the area under the curve (AUC) of the substance.
Thorough QT study (TQT)
Study objectives
A QT interval is a measure of cardiac rhythm. A QT interval can be measured using an electrocardiogram (ECG) and can be used as an (imperfect) biomarker to assess the risk that a medicine may provoke arrhythmia. In an ECG the activity of the electrical activity of the heart is measured and displayed as waves labelled as ‘P’, ‘Q’, ‘R’, ‘S’, and ‘T’. The QT interval is a measurement between the beginning of the Q wave and the end of the T wave.
Study design
TQT studies are conducted in vivo for all new molecular entities (NMEs). They must take place prior to Phase III trials, regardless of in vitro or non-clinical findings.
Typically, TQT studies are conducted as single dose crossover studies in healthy participants. Researchers evaluate therapeutic and supratherapeutic (greater than therapeutic) doses of the medicine against a positive control (such as a common antibiotic like moxifloxacin) and a negative control (a placebo).
A2-5.03.4-V1.1