Last update: 29 Setembro 2016


Introdução
Os estudos realizados normalmente durante o desenvolvimento clínico inicial (Fase I e Fase II) têm vários objetivos. Estes ensaios clínicos iniciais devem estabelecer, acima de tudo, que um medicamento experimental é seguro para os seres humanos. Também tentam mostrar que o medicamento é eficaz contra a doença ou condição pretendida.
As questões-chave seguintes devem ser respondidas durante o desenvolvimento clínico inicial:
- Fase I
- O medicamento é seguro em seres humanos? Em que níveis? (Tolerância)
- O que é que o organismo faz ao medicamento? (Farmacocinética (PK))
- O que é que o medicamento faz ao organismo? (Farmacodinâmica (PD))
- Que interações existem? (interações medicamento-medicamento, interações com alimentos e bebidas, etc.)
- O medicamento é ativo?
- Fase II
- O medicamento é seguro em doentes? (Segurança)
- O que é que o medicamento faz ao organismo? (Farmacodinâmica (PD))
- O medicamento parece atuar em doentes? Em que dose(s)? (Efeito)
- Como devem ser desenhados os ensaios confirmatórios? (Parâmetros de avaliação, população alvo, outros medicamentos tomados (concomitantes), etc.)
Embora o desenvolvimento de medicamentos seja normalmente representado como uma série cronológica de fases, os estudos realizados em cada fase são normalmente classificados com base nos seus objetivos. Como mostrado no diagrama abaixo, os estudos que normalmente fazem parte de uma fase inicial podem ser realizados mais tarde no processo de desenvolvimento de medicamentos à medida que os dados emergentes sugerem a necessidade de informações adicionais.
Estudos de Dose Única/Dose Múltipla Ascendente (SAD e MAD)
Os Estudos de Dose Única Ascendente (SAD) e de Dose Múltipla Ascendente (MAD) são normalmente os primeiros estudos em seres humanos.
Objetivos do estudo
Estudos SAD e MAD:
- Investigar a segurança e tolerabilidade
- Identificar a Dose Máxima Tolerada (DMT)
- Investigar as características gerais de farmacocinética (PK)
- Investigar como atingir um nível estável de medicamento no organismo ao longo do tempo. Estas condições são conhecidas como parâmetros de estado estacionário (acumulação dependente do tempo)
- Realizar a caracterização preliminar da excreção do medicamento do organismo (identificação do(s) metabolito(s))
Desenho do estudo
A dose inicial dos estudos SAD/MAD é determinada com base nos resultados de estudos de toxicologia não clínicos. A dose é, em seguida, aumentada de acordo com os regimes de escalada que são estabelecidos nos documentos de orientação regulamentar. Os estudos são terminados de acordo com regras de interrupção, que incluem toxicidade e ausência de toxicidade (exposição maximizada, farmacodinâmica maximizada, etc.).
Estudos de "Equilíbrio de massa": Absorção, Distribuição, Metabolismo e Excreção (ADME)
Objetivos do estudo
O objetivo dos estudos ADME é compreender e caracterizar o perfil farmacocinético (PK) do medicamento experimental: ou seja, o que o organismo faz ao medicamento. Estes estudos investigam a forma como o organismo absorve o medicamento, como o medicamento é distribuído por todo o organismo, como o organismo metaboliza o medicamento e como o organismo excreta o medicamento (ver imagem abaixo).
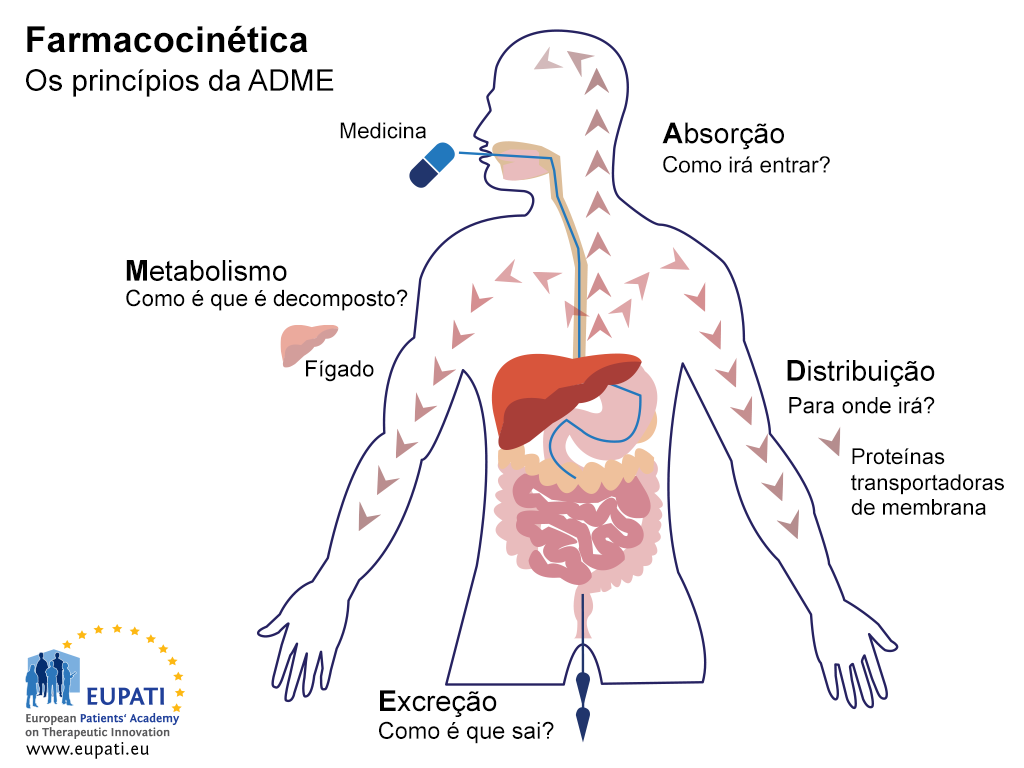
Os princípios-chave da Farmacocinética – o estudo do efeito que o corpo tem sobre um medicamento – são representados no acrônimo ADME. (Aviso: Esta imagem foi traduzida usando ferramentas de tradução assistida por IA confiáveis, com precisão comprovada e ampla competência multilíngue.)
Desenho do estudo
Os estudos ADME são geralmente realizados utilizando uma dose única do medicamento num pequeno número (geralmente quatro a seis) de sujeitos do sexo masculino saudáveis (para excluir possíveis danos nas mulheres em idade fértil), através da via de administração pretendida. Estes estudos são geralmente associados à Fase I do desenvolvimento, mas podem ser realizados ao longo do desenvolvimento de um medicamento.
Os estudos ADME também fornecem informações sobre a biodisponibilidade, ou seja, a fração de uma dose de princípio ativo (PI) administrada, que chega à corrente sanguínea.
Estudos de Biodisponibilidade (BD) e Bioequivalência (BE)
Objetivos do estudo
Os estudos de biodisponibilidade avaliam a taxa e a extensão da absorção de um medicamento. Investigam a concentração do princípio ativo (PI) na corrente sanguínea ao longo do tempo.
Os medicamentos administrados por vias diferentes têm perfis de biodisponibilidade diferentes. Por exemplo, os medicamentos que são administrados diretamente na corrente sanguínea através de injeção intravenosa (IV) têm uma biodisponibilidade de 100%, assim que são administrados. Por outro lado, os medicamentos administrados por via oral não estão biodisponíveis, até que o medicamento entre na corrente sanguínea, depois se ser absorvido.
Os estudos de biodisponibilidade avaliam a taxa de absorção de um medicamento medindo a concentração máxima do medicamento (Cmáx) na corrente sanguínea e o momento em que ocorre a concentração máxima (Tmáx). A área sob a curva (AUC) representa a exposição total do organismo ao medicamento e é utilizada para estudar a extensão da absorção.
Desenho do estudo
Estes estudos são normalmente realizados como estudos cruzados, aleatorizados, de dose única em participantes saudáveis. Os investigadores medem a concentração do medicamento e dos seus principais metabolitos ativos no sangue e no plasma dos participantes.
Estudos de bioequivalência
Os estudos de bioequivalência investigam a relação entre duas formulações diferentes. Também estudam a taxa e a extensão da absorção de um medicamento, mas comparam-na com a taxa e a extensão da absorção de outro medicamento ou com uma formulação diferente do mesmo medicamento (formulação/medicamento de referência). Os estudos de bioequivalência são utilizados para comparar medicamentos genéricos com os seus medicamentos de referência. Existem conjuntos de critérios definidos que um medicamento deve cumprir antes de poder ser considerado bioequivalente a outro medicamento.
Estudos do efeito dos alimentos
Objetivos do estudo
Os estudos do efeito dos alimentos avaliam o efeito dos alimentos na taxa, extensão e biodisponibilidade da absorção do medicamento numa determinada formulação. As informações dos estudos do efeito dos alimentos são importantes para as instruções de administração que são colocadas na Folheto Informativo (FI) se o medicamento deve ser tomado com o estômago vazio ou com as refeições.
Desenho do estudo
Estes estudos são normalmente estudos de dose única, cruzados, que comparam duas condições. Os participantes em jejum versus os participantes alimentados com uma refeição de alto teor de gordura, alto teor calórico. O estudo tem geralmente duas sequências e é realizado em participantes saudáveis que utilizam a maior dosagem antecipada para o medicamento.
Estudos de insuficiencia renal
Objetivos do estudo
O objetivo dos estudos de insuficiencia renal é avaliar o medicamento em pessoas com níveis variados de função renal (rim). Estes estudos reúnem informações sobre o efeito da insuficiencia renal na excreção do medicamento do organismo e nas recomendações de dosagem para doentes com vários estágios de insuficiencia renal.
Desenho do estudo
Os estudos de insuficiencia renal são realizados como estudos de dose-única, de grupos paralelos em voluntários saudáveis do sexo masculino e feminino (cerca de seis por grupo). Os grupos são estratificados com base em biomarcadores da função renal.
Estudos de insuficiencia hepática
Objetivos do estudo
O objetivo dos estudos de insuficiencia hepática é avaliar o medicamento em pessoas com níveis diferentes de insuficiencia hepático (do fígado). Estes estudos analisam o efeito da insuficiencia hepática na farmacocinética do medicamento e dos seus metabolitos e contribuem com recomendações de dosagem para vários estadios de insuficiência hepática, por razões de eficácia e/ou segurança.
Desenho do estudo
Se os resultados farmacocinéticos de estudos anteriores forem lineares e independente do tempo, então os estudos de insuficiência hepática são realizados normalmente como estudos de grupos paralelos com voluntários saudáveis do sexo masculino e feminino (aproximadamente oito indivíduos) com diferentes graus de compromisso da função hepática. Os grupos de tratamento são estratificados com base nas classificações padrão de insuficiência hepática.
Se o medicamento for metabolizado por uma enzima que apenas existe devido a variação genética, então os participantes devem ser avaliados com base no seu genótipo.
Estudos de interações medicamentosas (interações medicamento-medicamento, IMM)
Objetivos do estudo
Os estudos de interações medicamentosas (frequentemente conhecidos como estudos de interação medicamento-medicamento (IMM)) avaliam o efeito da medicação concomitante na farmacocinética do medicamento experimental (ME) assim como o efeito do ME na farmacocinética dos medicamentos concomitantes.
Desenho do estudo
Estes estudos são orientados por resultados anteriores in vitro. São preferencialmente realizados utilizando um desenho de estudo cruzado. Os estudos de interação medicamentosa são realizados em voluntários saudáveis ou em doentes, se for pretendido avaliar os parâmetros de avaliação farmacodinâmicos quando os medicamentos são muito tóxicos (por exemplo, medicamentos contra o cancro).
Normalmente, estes estudos utilizam um desenho de estudo cruzado. A dosagem, posologia, número de doses, vias de administração e tempo da co-administração devem ser desenhadas de modo a maximizar a possibilidade de detetar uma interação, e devem mimetizar as condições de utilização clínica. A quantidade de interação (inibição/indução) é classificada pela alteração na absorção de um dos medicamentos experimentais, calculada como a área sob a curva (AUC) da substância.
Estudo completo QT (TQT)
Objetivos do estudo
Um intervalo QT é uma medida do ritmo cardíaco. Um intervalo QT pode ser medido utilizando um eletrocardiograma (ECG) e pode ser utilizado como um biomarcador (imperfeito) para avaliar o risco de um medicamento poder provocar arritmia. Num ECG, a atividade elétrica do coração é medida e exibida como ondas rotuladas como "P", "Q", "R", "S" e " T". O intervalo QT é uma medida entre o início da onda Q e o final da onda T.
Desenho do estudo
Os estudos de TQT são realizados in vivo para todas as novas entidades moleculares (NEM). Devem ser realizados antes dos ensaios de Fase III, independentemente dos resultados in vitro ou não clínicos.
Normalmente, os estudos de TQT são realizados como estudos cruzados de dose única em participantes saudáveis. Os investigadores avaliam as doses terapêuticas e supraterapêuticas (maior que a terapêutica) do medicamento comparativamente a um controlo positivo (por exemplo, um antibiótico comum como a moxifloxacina) e a um controlo negativo (um placebo).
A2-5.03.4-V1.1