Last update: 29 september 2016


Introduktion
De undersøgelser, der typisk gennemføres tidligt i klinisk udvikling (fase I og fase II), har flere forskellige formål. Disse tidlige kliniske forsøg skal først og fremmest påvise, at et testlægemiddel er sikkert for mennesker. De skal også forsøge at vise, at lægemidlet er effektivt mod den tilsigtede sygdom eller tilstand.
Følgende vigtige spørgsmål skal besvares tidligt i klinisk udvikling:
- Fase I
- Er lægemidlet sikkert for mennesker? Ved hvilke niveauer? (Tolerance)
- Hvad gør kroppen ved lægemidlet? (Farmakokinetik)
- Hvad gør lægemidlet ved kroppen? (Farmakodynamik)
- Hvilke interaktioner er der? (Interaktioner med andre lægemidler, interaktioner med mad og drikke osv.)
- Er lægemidlet aktivt?
- Fase II
- Er lægemidlet sikkert for patienterne? (Sikkerhed)
- Hvad gør lægemidlet ved kroppen? (Farmakodynamik)
- Ser det ud til, at lægemidlet virker hos patienterne? Ved hvilke doser? (Effekt)
- Hvordan skal designet af bekræftende forsøg være? (Endepunkter, målgruppe, brug af andre lægemidler (samtidig) osv.)
Selvom udvikling af lægemidler typisk fremstilles som en kronologisk række af faser, klassificeres de undersøgelser, der udføres i de forskellige faser, typisk efter deres formål. Som vist i diagrammet nedenfor kan undersøgelser, der typisk indgår i en tidligere fase, udføres senere i udviklingsprocessen for lægemidler, når nye data fremkommer, som viser et behov for yderligere oplysninger.
-
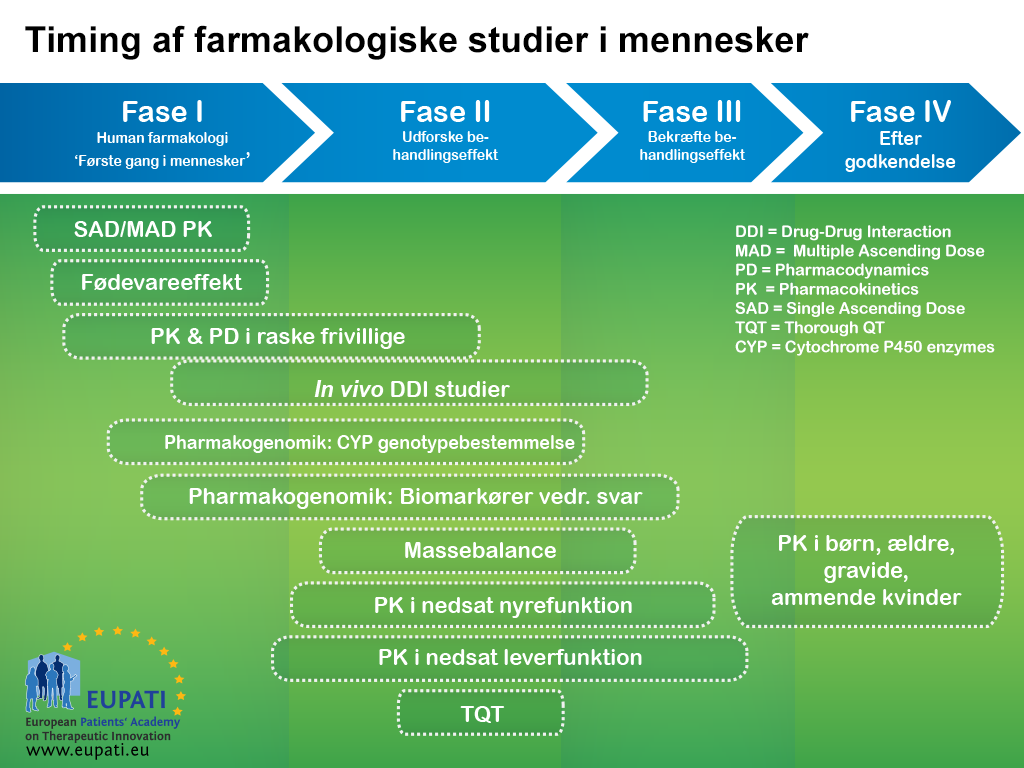
- Logikken bag præsentationen af lægemiddeludvikling som et antal faser i en bestemt rækkefølge kommer fra en idé om, at resultaterne fra forudgående undersøgelser bør have indflydelse på planerne for efterfølgende undersøgelser: Nye data giver ofte anledning til ændringer i udviklingsstrategierne.
Eskaleringsundersøgelser af enkelt stigende dosis/flere stigende doser (SAD – Single Ascending Dose og MAD – Multiple Ascending Dose)
Undersøgelser af enkelt stigende dosis (SAD – Single Ascending Dose) og flere stigende doser (MAD – Multiple Ascending Dose) er som regel de første, der involverer mennesker.
Undersøgelsesformål
SAD- og MAD-undersøgelser:
- At undersøge sikkerhed og tolerabilitet
- At bestemme en maksimalt tolereret dosis (MTD)
- At undersøge generelle farmakokinetiske karakteristika
- At undersøge, hvordan der opnås et stabilt lægemiddelniveau i kroppen over tid. Disse forhold kaldes steady-state-parametre (akkumuleringstid-afhængighed)
- At foretage foreløbig undersøgelse af lægemidlets udskillelse fra kroppen (og bestemme metabolitter)
Undersøgelsesdesign
Startdosen i SAD/MAD-undersøgelser fastlægges på basis af resultaterne fra de non-kliniske toksikologiundersøgelser. Dosen øges derpå efter en eskaleringsplan, som findes i vejledende dokumenter fra lægemiddelmyndighederne. Undersøgelserne stopper i henhold til stopreglerne, der omfatter toksicitet og fravær af toksicitet (maksimeret eksponering, maksimeret farmakodynamik osv.).
Massebalanceundersøgelser: Absorption, fordeling, metabolisme og udskillelse (ADME – Absorption, Distribution, Metabolism, Excretion)
Undersøgelsesformål
Formålet med ADME-undersøgelser er at forstå og karakterisere den farmakokinetiske profil for forsøgslægemidlet: dvs. hvad kroppen gør ved lægemidlet. Disse undersøgelser ser på den måde, som kroppen absorberer lægemidlet på, hvordan lægemidlet fordeles i kroppen, hvordan kroppen metaboliserer lægemidlet, og hvordan kroppen udskiller lægemidlet (se billedet nedenfor).
-
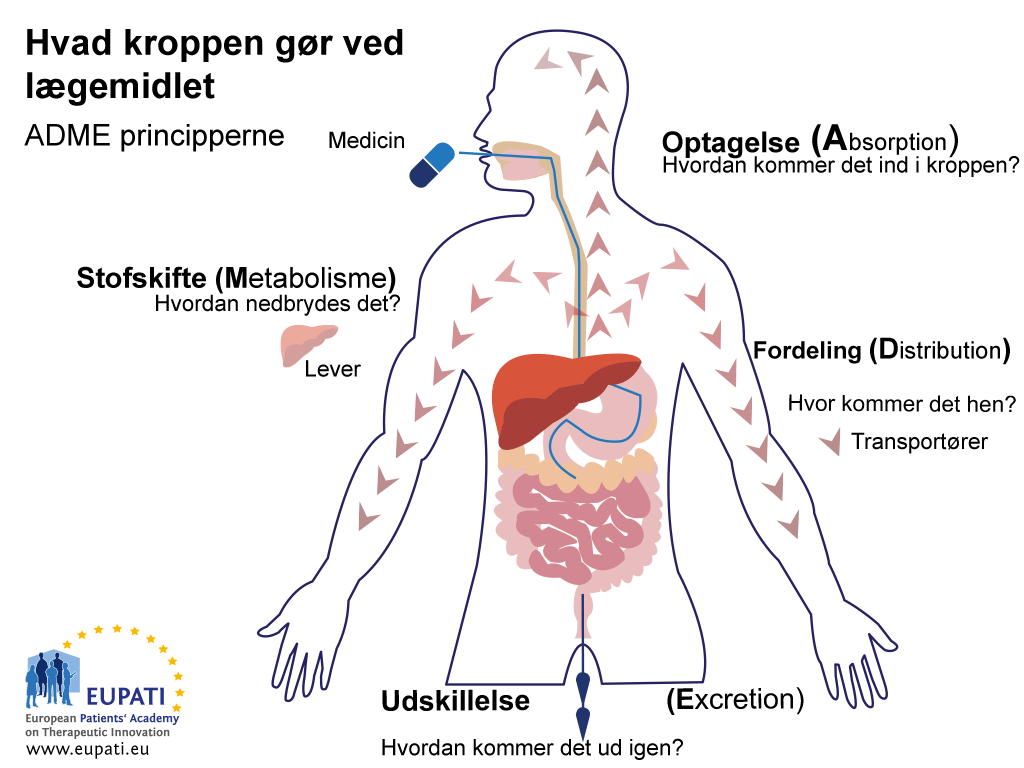
- Nøgleprincipperne i farmakokinese: studiet af kroppens indvirkning på medicin repræsenteres ved forkortelsen ADME.
Undersøgelsesdesign
ADME-undersøgelser udføres normalt med en enkelt dosis af lægemidlet hos et lille antal (som regel fire til seks) raske mænd (for at udelukke mulig skade hos kvinder i den fertile alder), via den tilsigtede administrationsvej. Disse undersøgelser er typisk forbundet med fase I-udvikling, men de kan gennemføres i hele udviklingsprocessen for lægemidlet.
ADME-undersøgelser giver oplysninger om biotilgængelighed – dvs. den brøkdel af en indgivet aktiv farmaceutisk ingrediens (API – active pharmaceutical ingredient), der når frem til blodbanen.
Biotilgængeligheds- og bioækvivalensundersøgelser
Undersøgelsesformål
I biotilgængelighedsundersøgelser vurderes hastigheden og omfanget af absorptionen af et lægemiddel. Koncentrationen af den aktive farmaceutiske ingrediens (API) i blodbanen undersøges over tid.
Lægemidler, der indgives ad forskellige veje, har forskellige biotilgængelighedsprofiler. For eksempel har lægemidler, der indgives direkte i blodbanen via en intravenøs injektion (iv), en biotilgængelighed på 100 %, straks når de indgives, mens lægemidler, der indgives gennem munden, ikke er biotilgængelige, før lægemidlet kommer ind i blodbanen, efter at det er blevet absorberet.
-
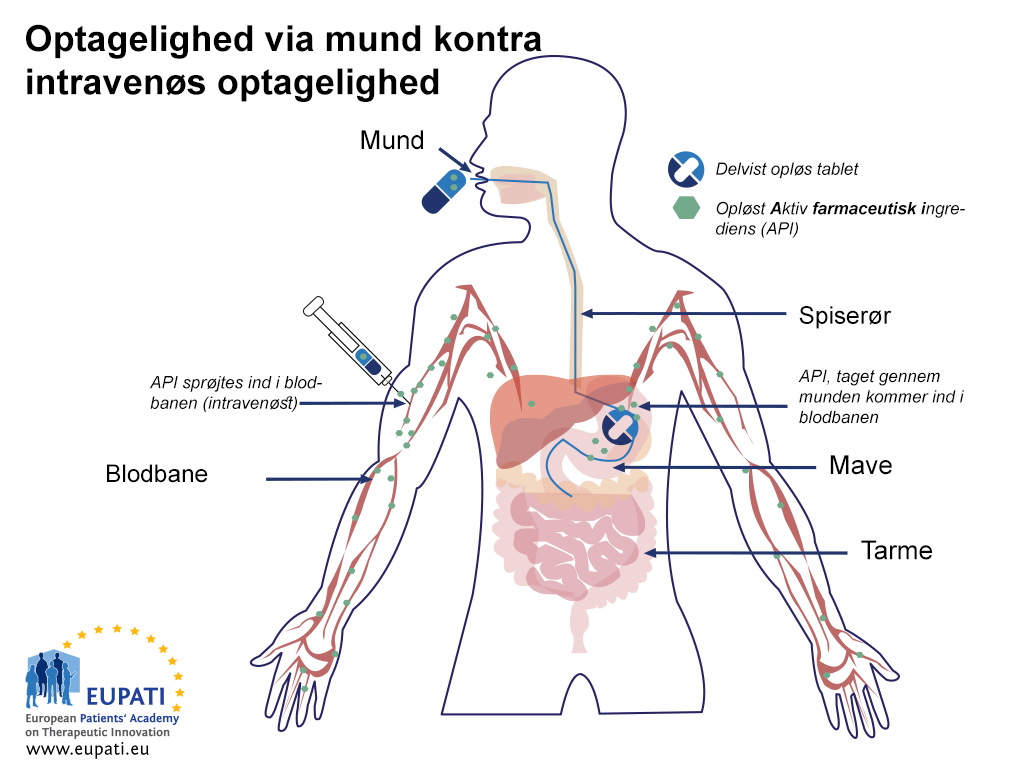
- Sammenligning af biotilgængeligheden for aktive farmaceutiske ingredienser, der indgives oralt og intravenøst. Det siges, at en aktiv farmaceutisk ingrediens er “biologisk tilgængelig” (biotilgængelig), når den kommer ind i blodet.
I biotilgængelighedsundersøgelser vurderes absorptionshastigheden for et lægemiddel ved at måle den maksimale koncentration af lægemidlet (Cmax) i blodbanen og det tidspunkt, hvor den maksimale koncentration forekommer (Tmax). Arealet under kurven (AUC) repræsenterer kroppens samlede eksponering for lægemidlet og bruges til at undersøge omfanget af absorption.
-
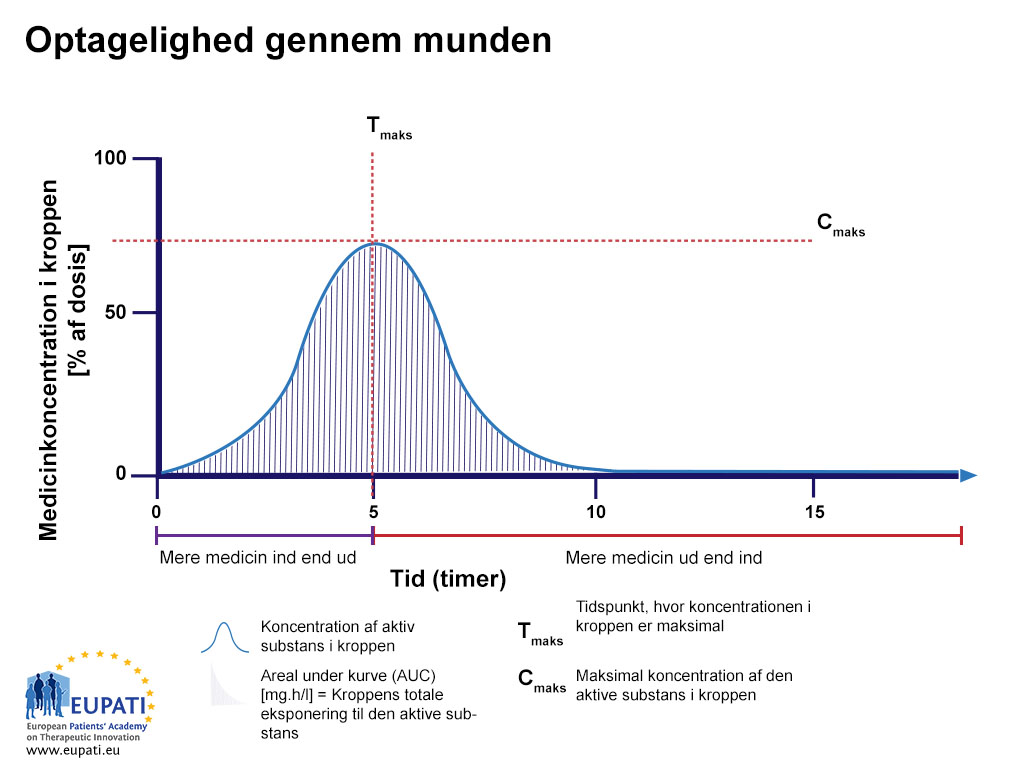
- Procentdelen af aktivt stof, efter en tablet er blevet slugt, undersøgt i en periode på 15 timer. Arealet under kurven er skraveret og repræsenterer den samlede mængde aktivt stof, som var i blodet i løbet af den undersøgte periode. Tmax er det tidspunkt, hvor den højeste koncentration af lægemidlet findes i blodet, hvorimod Cmax er den maksimale koncentration af lægemidlet, som findes i blodet.
Undersøgelsesdesign
Disse undersøgelser gennemføres typisk som randomiserede, enkeltdosis-overkrydsningsforsøg med raske deltagere. Forskerne måler koncentrationen af lægemidlet og dets vigtigste aktive metabolitter i deltagernes blod og plasma.
Bioækvivalensundersøgelser
I bioækvivalensundersøgelser ser forskerne på forholdet mellem to forskellige formuleringer. De undersøger også hastigheden og omfanget af absorptionen af et lægemiddel, men de sammenligner dette med hastigheden og omfanget af absorptionen af en anden formulering af det samme lægemiddel (referenceformulering/lægemiddel). Bioækvivalensundersøgelser bruges til at sammenligne generiske lægemidler med referencelægemidlerne. Der er nogle faste kriterier, som et lægemiddel skal opfylde, før det kan anses for at være bioækvivalent med et andet lægemiddel.
Undersøgelser af effekten af fødevarer
Undersøgelsesformål
I undersøgelser af fødevarers effekt vurderes effekten af fødevarer på hastigheden, omfanget og biotilgængeligheden af et lægemiddels absorption i en given formulering. Oplysninger fra undersøgelser af fødevarers effekt er vigtige for de administrationsanvisninger, der gives i indlægssedlen, og som angiver, om lægemidlet skal tages på tom mave eller ved et måltid.
Undersøgelsesdesign
Disse undersøgelser er typisk enkeltdosis-overkrydsningsundersøgelser, der sammenligner to situationer: deltagere, der har fastet, i forhold til deltagere, der har fået et fedtholdigt, kalorierigt måltid. Undersøgelsen har normalt to sekvenser og udføres med raske deltagere og brug af den højeste forventede styrke af lægemidlet.
Undersøgelser af nedsat nyrefunktion
Undersøgelsesformål
Formålet med undersøgelser af nedsat nyrefunktion er at vurdere lægemidlet hos personer med forskelligt nyrefunktionsniveau. I disse undersøgelser indsamles oplysninger om effekten af nedsat nyrefunktion på udskillelsen af lægemiddel fra kroppen og dosisanbefalinger til patienter med forskellige stadier af nedsat nyrefunktion.
Undersøgelsesdesign
Undersøgelser af nedsat nyrefunktion gennemføres som enkeltdosis-parallelgruppeforsøg med raske mandlige og kvindelige frivillige (omkring seks personer pr. gruppe). Grupperne stratificeres på basis af biomarkører for nyrefunktion.
Undersøgelser af nedsat leverfunktion
Undersøgelsesformål
Formålet med undersøgelser af nedsat leverfunktion er at vurdere lægemidlet hos personer med forskellige niveauer af nedsat leverfunktion. I disse undersøgelser ser forskerne på effekten af nedsat leverfunktion på lægemidlets og dets metabolitters farmakokinetik og giver dosisanbefalinger til forskellige stadier af nedsat leverfunktion af virknings- og/eller sikkerhedsårsager.
Undersøgelsesdesign
Hvis de farmakokinetiske resultater fra tidligere undersøgelser er lineære og tidsuafhængige, gennemføres undersøgelser af nedsat leverfunktion typisk som parallelgruppeundersøgelser med raske mandlige og kvindelige frivillige (omkring otte personer) med forskellige grader af nedsat leverfunktion. Behandlingsgrupperne stratificeres på basis af standardklassificeringer af nedsat leverfunktion.
Hvis lægemidlet metaboliseres af et enzym, der kun forekommer på grund af en genetisk variation, skal deltagerne vurderes ud fra deres genotypestatus.
Undersøgelser af interaktioner med andre lægemidler (lægemiddelinteraktion)
Undersøgelsesformål
I undersøgelser af lægemiddelinteraktion evalueres effekten af lægemidler, der bruges samtidig, på forsøgslægemidlets farmakokinetik samt effekten af forsøgslægemidlet på de lægemidler, der bruges samtidig.
Undersøgelsesdesign
Disse undersøgelser baseres på tidligere in vitro-resultater, og de udføres fortrinsvis med et overkrydsningsforsøgsdesign. Undersøgelser af lægemiddelinteraktion udføres med raske frivillige eller med patienter, hvis farmakodynamiske endepunkter skal kunne vurderes, når lægemidlerne er for toksiske (f.eks. lægemidler mod kræft).
Til disse undersøgelser bruges typisk et forsøgsdesign med overkrydsningsforsøg. Dosering, doseringsintervaller, antal doser, administrationsveje og tidspunkt for samtidig administration skal tilrettelægges, så muligheden for at påvise en interaktion maksimeres, og så det kliniske regi efterlignes. Mængden af interaktion (inhibition/induktion) klassificeres af ændringen i absorptionen af ét af forsøgslægemidlerne, beregnet som arealet under kurven (AUC) for stoffet.
TQT-undersøgelse (Thorough QT)
Undersøgelsesformål
QT-interval er et mål for hjerterytme. Et QT-interval kan måles ved hjælp af et elektrokardiogram (EKG) og kan bruges som en (ufuldstændig) biomarkør til vurdering af risikoen for, at et lægemiddel fremkalder arytmi. I et EKG måles hjertets elektriske aktivitet og vises som takker, der er markeret med ‘P’, ‘Q’, ‘R’, ‘S’ og ‘T’. QT-intervallet er en måling mellem begyndelsen af Q-takken og slutningen af T-takken.
Undersøgelsesdesign
TQT-undersøgelser gennemføres in vivo for alle nye molekyleelementer. De skal foretages forud for fase III-forsøg, uafhængigt af in vitro- eller non-kliniske resultater.
TQT-undersøgelser gennemføres typisk som enkeltdosis-overkrydsningsforsøg med raske deltagere. Forskerne vurderer terapeutiske og supraterapeutiske (større end terapeutiske) doser af lægemidlet i forhold til et positivt kontrolpræparat (f.eks. et almindeligt antibiotikum som moxifloxacin) og et negativt kontrolpræparat (placebo).
A2-5.03.4-V1.1