Last update: 29 Settembre 2016


Introduzione
Gli studi condotti di solito nel corso dello sviluppo clinico iniziale (Fase I e Fase II) comportano una varietà di obiettivi. Tali studi clinici iniziali devono stabilire soprattutto che un prodotto medicinale sperimentale sia sicuro per gli esseri umani. Cercano inoltre di provare a dimostrare che il farmaco sia efficace nei confronti della malattia o del disturbo previsti.
Durante lo sviluppo clinico iniziale, è necessario rispondere alle seguenti domande chiave:
- Fase I
- Il farmaco è sicuro in esseri umani? A quali livelli? (Tolleranza)
- In che modo l’organismo agisce sul farmaco? (Farmacocinetica (PK))
- In che modo il farmaco agisce sull’organismo? (Farmacodinamica (PD))
- Che interazioni si verificano? (Interazioni farmaco-farmaco, interazioni con cibo e bevande ecc.)
- Il farmaco è attivo?
- Fase II
- Il farmaco è sicuro nei pazienti? (Sicurezza)
- In che modo il farmaco agisce sull’organismo? (Farmacodinamica (PD))
- Il farmaco sembra funzionare sui pazienti? A quale/i dose/i? (Effetto)
- In che modo devono essere disegnati gli studi di conferma? (Endpoint, popolazione target, altri farmaci assunti (Concomitante) ecc.)
Sebbene lo sviluppo dei farmaci sia generalmente rappresentato come una serie cronologica di fasi, gli studi che sono effettuati entro ciascuna fase sono di solito classificati sulla base dei loro obiettivi. Come mostrato del diagramma riportato sotto, gli studi che in genere fanno parte della fase iniziale possono essere eseguiti successivamente nel processo di sviluppo dei farmaci quando i dati che emergono suggeriscono la necessità di informazioni aggiuntive.
Studi di progressione della dose singola/dose multipla crescente (SAD e MAD)
Gli studi della dose singola crescente (SAD) e della dose multipla crescente (MAD) sono di solito studi iniziali in esseri umani.
Obiettivi dello studio
Studi SAD e MAD:
- Indagano la sicurezza e la tollerabilità
- Individuano la dose massima tollerata (Maximum Tolerated Dose, MTD)
- Esaminano le caratteristiche generali di farmacocinetica (PK)
- Esaminano in che modo raggiungere un livello stabile di farmaco nell'organismo nel corso del tempo, tali condizioni sono note come parametri di stato stazionario (accumulo dipendente dal tempo).
- Effettuare un'esplorazione preliminare di escrezione del farmaco dall'organismo (identificazione del metabolita/dei metaboliti).
Disegno dello studio
La dose di partenza per gli studi SAD/MAD è determinata sulla base dei risultati di studi tossicologici non clinici. La dose viene poi aumentata secondo schemi di escalation che vengono esposti in documenti relativi alle linee guida normative. Gli studi vengono interrotti secondo regole, le quali comprendono tossicità e assenza di tossicità (esposizione massimizzata, farmacodinamica massimizzata ecc.).
Studi di "equilibrio della massa": assorbimento, distribuzione, metabolismo, escrezione (ADME)
Obiettivi dello studio
L'obiettivo degli studi ADME è quello di comprendere e caratterizzare il profilo di farmacocinetica (PK) del prodotto medicinale sperimentale: vale a dire, ciò che l'organismo fa al farmaco. Tali studi esaminano il modo in cui l'organismo assorbe il farmaco, come il medicinale viene distribuito in tutto l'organismo, come quest'ultimo metabolizza il farmaco e il modo in cui lo espelle (vedere l'immagine riportata sotto).
-
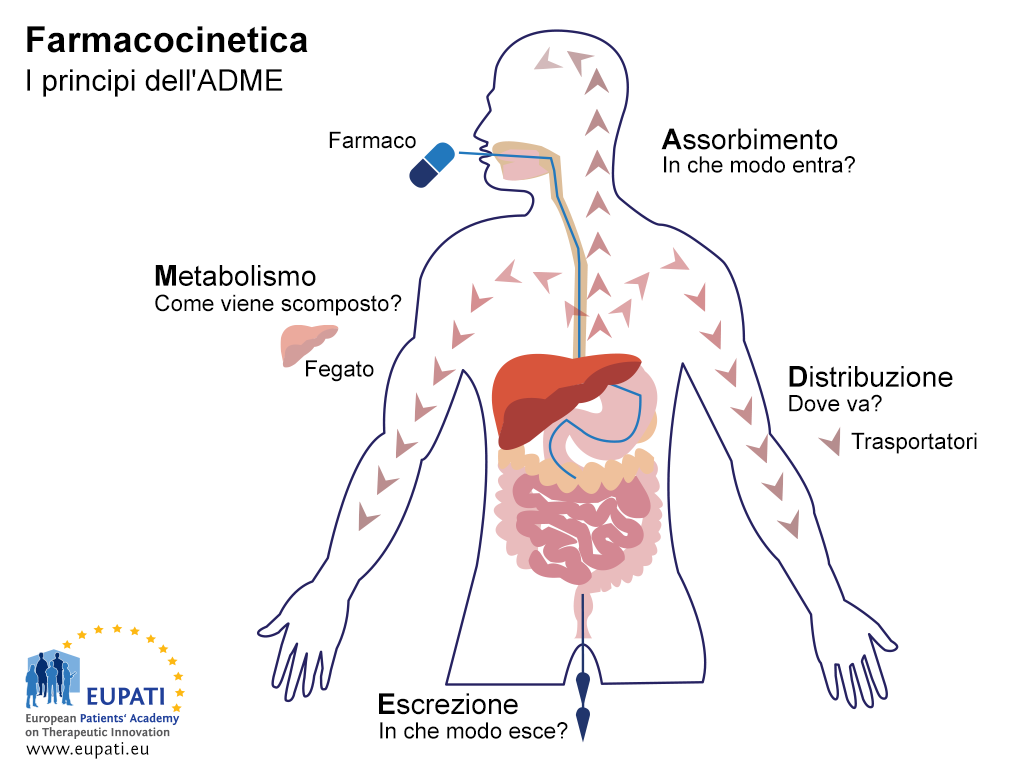
- I principi fondamentali della farmacocinetica, cioè lo studio dell’effetto prodotto dall’organismo su un farmaco, sono rappresentati nell’acronimo ADME.
Disegno dello studio
Gli studi ADME sono di solito effettuati utilizzando una dose singola del farmaco in un piccolo numero (di solito da quattro a sei) di maschi sani (al fine di escludere danni a donne in età fertile), tramite la via di somministrazione desiderata. Tali studi sono generalmente associati alla Fase I dello sviluppo, ma possono essere eseguiti nel corso dello sviluppo di un prodotto medicinale.
Gli studi ADME forniscono anche informazioni sulla biodisponibilità, vale a dire la frazione di una dose somministrata di principio attivo (active pharmaceutical ingredient, API) che raggiunge il flusso sanguigno.
Studi di biodisponibilità (BA) e bioequivalenza (BE)
Obiettivi dello studio
Gli studi di biodisponibilità valutano la frequenza e l'entità dell'assorbimento di un farmaco. Studiano la concentrazione del principio attivo (API) nel flusso sanguigno nel corso del tempo.
I farmaci somministrati tramite vie diverse hanno profili differenti di biodisponibilità. Ad esempio, i farmaci che vengono somministrati direttamente nel flusso sanguigno tramite iniezione endovenosa (intravenous injection, IV) hanno una disponibilità del 100% appena vengono somministrati, mentre i medicinali che vengono somministrati per via orale fino a che il farmaco entra nel flusso sanguigno dopo il suo assorbimento da parte dell'organismo.
-
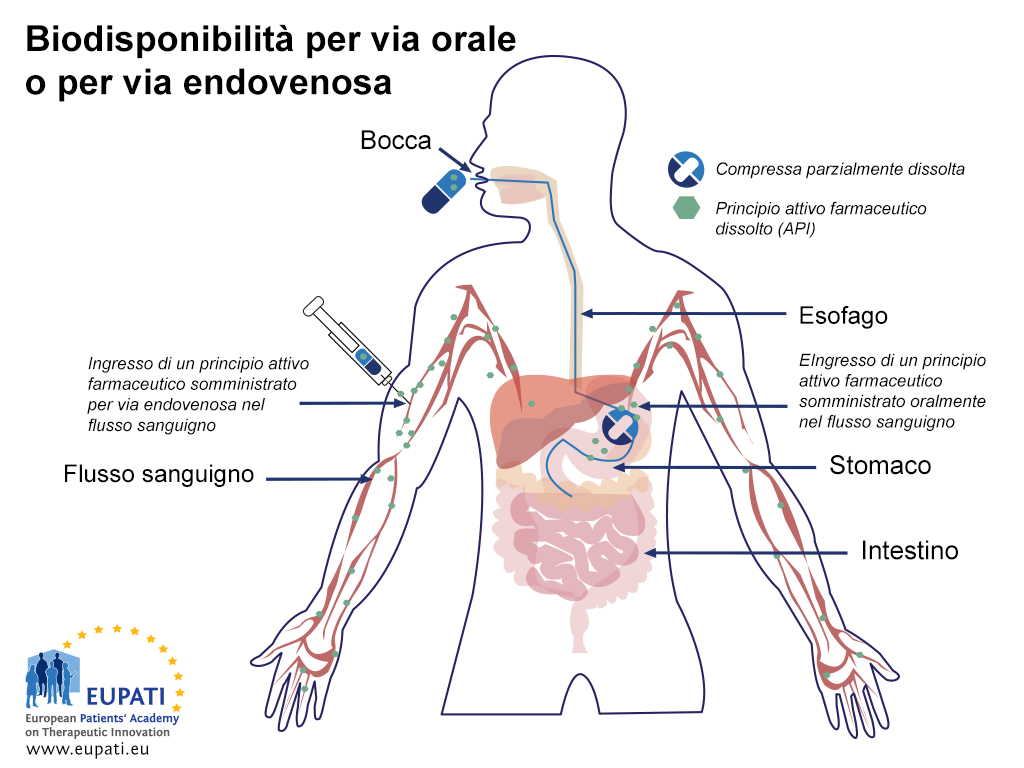
- Immagine schematica che mostra l’assorbimento di una capsula assunta per via orale rispetto all’iniezione di un farmaco praticata direttamente nel flusso sanguigno (iniezione endovenosa). Dopo aver raggiunto lo stomaco, la capsula viene ulteriormente trasportata all’intestino tenue dove viene ulteriormente assorbita.
Gli studi di biodisponibilità valutano il tasso di assorbimento di un farmaco misurando la concentrazione massima del farmaco (Cmax) nel flusso sanguigno e il tempo in cui si verifica la concentrazione massima (Tmax). L'area sotto al curva (area under the curve, AUC) rappresenta l'esposizione totale dell'organismo al farmaco e viene utilizzata per studiare l'entità dell'assorbimento.
-
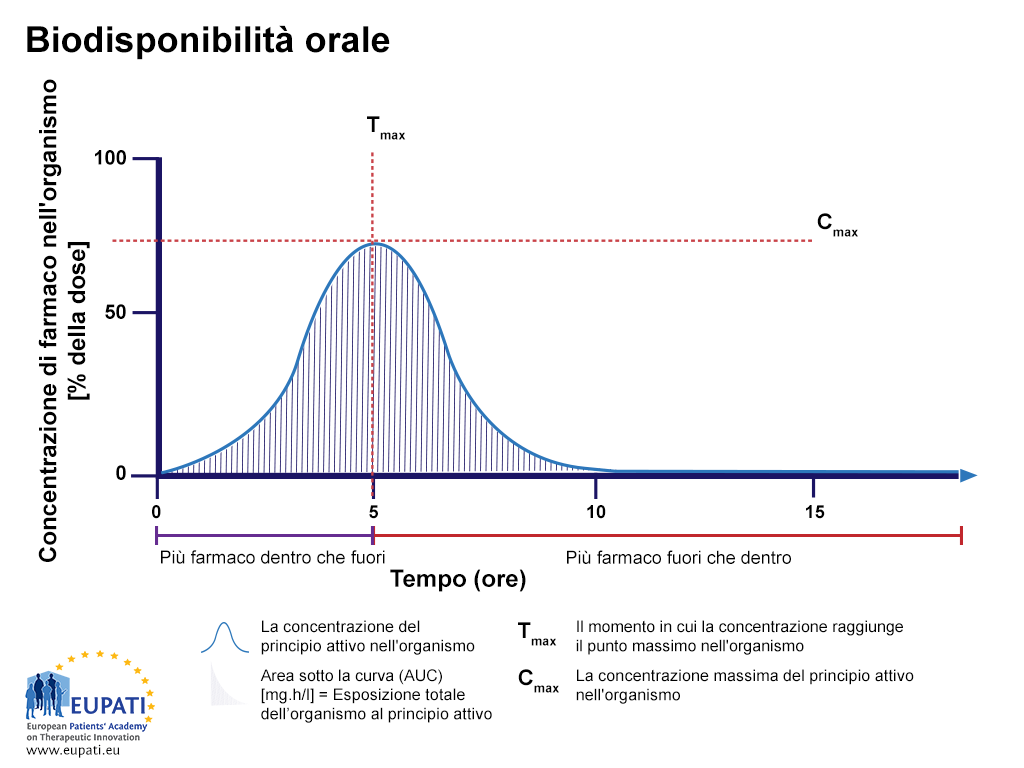
- La percentuale di farmaco attivo dopo che una compressa viene inghiottita, studiata per un periodo di 15 ore. AUC è la parte colorata. Il momento in cui si riscontra la massima concentrazione di farmaco nel flusso sanguigno si chiama Tmax, mentre la concentrazione massima di farmaco rilevata nel flusso sanguigno si chiama Cmax.
Disegno dello studio
Tali studi vengono generalmente condotti come studi in crossover, randomizzati, a dose singola, in pazienti sani. I ricercatori misurano la concentrazione del farmaco e del suo principale metabolita attivo nel sangue e nel plasma dei partecipanti.
Studi di bioequivalenza
Gli studi di bioequivalenza indagano sulla relazione tra due diverse formulazioni. Questi inoltre studiano il tasso e l'entità dell'assorbimento di un farmaco, ma li confrontano con il tasso e con l'entità dell'assorbimento di altri farmaci o di una formulazione differente dello stesso farmaco (formulazione/farmaco di riferimento). Gli studi di bioequivalenza vengono utilizzati per confrontare farmaci generici con i loro farmaci di riferimento. Esistono criteri fissati che un farmaco deve soddisfare prima che possa essere considerato bioequivalente con un altro farmaco.
Studi sugli effetti degli alimenti
Obiettivi dello studio
Gli studi sugli effetti degli alimenti valutano l'effetto degli alimenti sul tasso, l'entità e la biodisponibilità del farmaco in una data formulazione. Le informazioni derivanti dagli studi sugli effetti degli alimenti sono importanti per le istruzioni relative alla somministrazione che vengono esposte nel foglio illustrativo (FI), le quali indicano se il farmaco deve essere preso a stomaco vuoto o in occasione dei pasti.
Disegno dello studio
Tali studi sono generalmente studi a dose singola, in crossover, che confrontano due condizioni: i partecipanti che digiunano rispetto ai partecipanti che vengono nutriti con un pasto ad alto indice di grassi e calorie. Lo studio di solito ha due sequenze e viene eseguito in partecipanti sani utilizzando la forza più elevata prevista del farmaco.
Studi sulla compromissione renale
Obiettivi dello studio
L'obiettivo degli studi di compromissione renale è di valutare il farmaco in individui con vari livelli di funzione renale. Tali studi raccolgono informazioni sull'effetto della compromissione renale sull'escrezione del farmaco dall'organismo e raccomandazioni di dosaggio per pazienti con diversi stadi di compromissione renale.
Disegno dello studio
Gli studi sulla compromissione renale vengono svolti come studi a dose singola in gruppi paralleli in volontari sani di sesso maschile e femminile (circa sei per gruppo). I gruppi vengono stratificati sulla base dei biomarcatori della funzione renale.
Studi sulla compromissione epatica
Obiettivi dello studio
L'obiettivo degli studi di compromissione epatica è di valutare il farmaco in individui con diversi livelli di compromissione epatica (del fegato). Tali studi esaminano l'effetto della compromissione epatica sulla farmacocinetica del farmaco e dei suoi metaboliti e forniscono raccomandazioni di dosaggio per vari stadi della compromissione epatica per motivi di efficacia e/o sicurezza.
Disegno dello studio
Se i risultati di farmacocinetica di precedenti stadi sono lineari e dipendenti dal tempo, gli studi di compromissione epatica sono generalmente svolti come studi su gruppi paralleli di volontari sani di sesso maschile e femminile (circa otto individui) con diversi gradi di compromissione della funzione epatica. I gruppi di trattamento vengono stratificati in base a classificazioni standard della compromissione epatica.
Se il farmaco viene metabolizzato da un enzima che è presente solo a causa di una variazione genetica, i partecipanti devono essere valutati conseguentemente sulla base del loro status di genotipo.
Studi di interazione farmaco-farmaco (DDI)
Obiettivi dello studio
Gli studi di interazione tra farmaci (noti comunemente come studi di interazione farmaco-farmaco (Drug-Drug Interaction, DDI)) stimano l'effetto di un farmaco concomitante sulla farmacocinetica del prodotto medicinale sperimentale (investigational medicinal product, IMP) così come l'effetto dell'IMP sulla farmacocinetica di farmaci concomitanti.
Disegno dello studio
Questi studi ricevono un orientamento da parte di precedenti risultati in vitro; sono preferibilmente condotti utilizzando un disegno di studio in crossover. Gli studi di interazione farmaco-farmaco vengono condotti in volontari sani o in pazienti se è desiderabile valutare gli endpoint di farmacodinamica quando i farmaci sono troppo tossici (ad esempio, farmaci anti-cancro).
Generalmente, tali studi utilizzano un disegno di studio in crossover. Il dosaggio, gli intervalli di dosaggio, il numero di dosi, le vie di somministrazione e le tempistiche di co-somministrazione devono essere pianificati per massimizzare la possibilità di individuare un'interazione e devono mimare un ambiente clinico. La quantità di interazioni (inibizione/induzione) viene classificata dalla variazione nell'assorbimento di uno dei prodotti medicinali sperimentali, calcolato come area sotto la curva (area under the curve, AUC) della sostanza.
Studio approfondito sul QT (TQT)
Obiettivi dello studio
Un intervallo di QT è una misura della frequenza cardiaca. Un intervallo di QT può essere misurato utilizzando un elettrocardiogramma (ECG) e può essere utilizzato come un biomarcatore (imperfetto) per valutare il rischio che un farmaco possa provocare aritmia. In un ECG l'attività elettrica del cuore viene misurata e indicata sotto forma di onde classificate come "P", "Q", "R", "S" e "T". L'intervallo di QT è una misurazione tra l'inizio dell'onda Q e la fine dell'onda T.
Disegno dello studio
Gli studi di TQT sono svolti in vivo per tutte nuove entità molecolari (new molecular entities, NME). Devono avere luogo prima degli studi di Fase III, indipendentemente dai riscontri in vitro o preclinici.
In genere, gli studi di TQT vengono condotti come studi in crossover in pazienti sani. I ricercatori valutano le dosi terapeutiche e sovraterapeutiche (maggiori di quelle terapeutiche) del farmaco rispetto a un controllo positivo (come un comune antibiotico come la moxifloxacina) e un controllo negativo (un placebo).
A2-5.03.4-V1.1