

Introducere
Studiile desfășurate în mod normal în primele faze ale dezvoltării clinice (Faza I și Faza II) au diferite obiective. Aceste studii clinice timpurii trebuie să demonstreze, înainte de toate, că un produs medic prezintă siguranță pentru oameni. Acestea urmăresc, de asemenea, să arate că medicamentul este eficient în tratarea bolii sau afecțiunii țintă.
În primele faze ale dezvoltării clinice, trebuie găsite răspunsurile la următoarele întrebări:
- Faza I
- Medicamentul prezintă siguranță pentru oameni? La ce niveluri? (toleranța)
- Cum acționează corpul asupra medicamentului? (farmacocinetica (FC))
- Cum acționează medicamentul în corp? (farmacodinamica (FD))
- Ce interacțiuni există? (interacțiuni între medicamente sau interacțiuni cu alimentele, băuturile etc.)
- Medicamentul este activ?
- Faza II
- Medicamentul prezintă siguranță pentru pacienți? (siguranța)
- Cum acționează medicamentul în corp? (farmacodinamica (FD))
- Medicamentul pare să funcționeze la pacienți? La ce doze? (efectul)
- Cum trebuie structurate studiile de confirmare? (punctele finale, populația țintă, alte medicamente administrate (concomitent) etc.)
Deși dezvoltarea medicamentelor este reprezentată în mod normal ca serie cronologică de faze, studiile efectuate în fiecare fază sunt clasificate, de obicei, în funcție de obiectivele acestora. Așa cum se arată în diagrama de mai jos, studiile efectuate în mod normal într-o fază mai timpurie pot avea loc mai târziu în procesul de dezvoltare a medicamentelor dacă datele emergente indică necesitatea unor informații suplimentare.
Studiile de escaladare cu doză unică/cu doze multiple crescătoare (DUC și DMC)
Studiile cu doză unică crescătoare (DUC) și cele cu doze multiple crescătoare (DMC) sunt, în general, primele studii pe oameni.
Obiectivele studiului
Studiile DUC și DMC:
- Investighează siguranța și tolerabilitatea
- Identifică doza maximă tolerată (DMT)
- Investighează caracteristicile farmacocinetice (FC) generale
- Investighează modul în care poate fi obținut un nivel stabil în timp al medicamentului în corp; condițiile pentru acest obiectiv sunt numite parametri în stare stabilă (dependența între timp și acumulare).
- Efectuează explorarea preliminară a excreției medicamentului din corp (identificarea metaboliților)
Structura studiului
Doza de pornire pentru studiile DUC/DMC este determinată pe baza rezultatelor studiilor non-clinice de toxicologie. Doza crește apoi conform schemelor de escaladare, descrise în documentele de îndrumare emise de către autoritățile de reglementare. Studiile sunt oprite conform regulilor de oprire, care includ toxicitatea și toxicitatea absentă (expunerea maximizată, farmacodinamica maximizată etc.).
Studiile de „echilibru masic”: Absorbția, Distribuția, Metabolizarea, Excreția (ADME)
Obiectivele studiului
Obiectivul studiilor ADME este de a înțelege și caracteriza profilul farmacocinetic (FC) al produsului medicamentos investigat: mai exact, ce face corpul cu medicamentul. Aceste studii investighează modul în care corpul absoarbe medicamentul, modul în care medicamentul este distribuit în corp, modul în care corpul metabolizează medicamentul și modul în care corpul elimină medicamentul (a se vedea imaginea de mai jos).
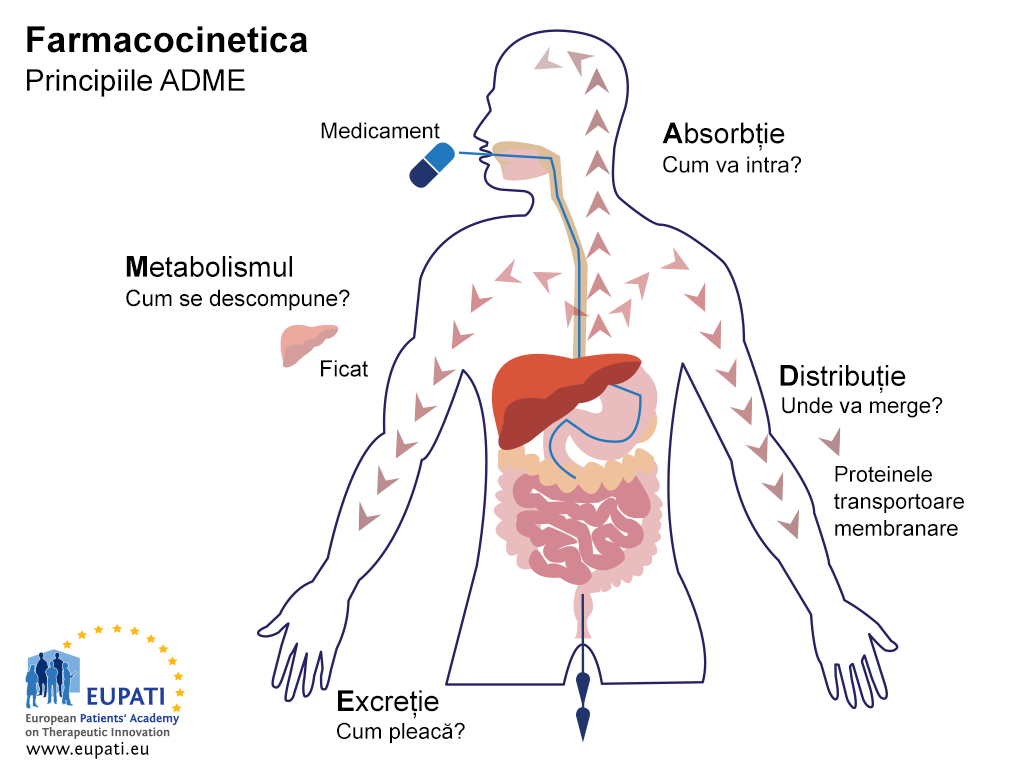
Principiile cheie ale Farmacocineticii - studiul efectului pe care corpul îl are asupra unui medicament - sunt reprezentate de acronimul ADME. (Avertisment: Această imagine a fost tradusă folosind instrumente de traducere asistate de inteligență artificială fiabile, cu o precizie dovedită și o competență multilingvă extinsă.)
Structura studiului
Studiile ADME sunt, în general, efectuate cu o singură doză a medicamentului, pe un număr mic (de la patru la șase) de bărbați sănătoși (pentru a se exclude eventualele riscuri la adresa femeilor cu potențial de sarcină), folosindu-se calea de administrare prevăzută. Aceste studii sunt, în general, asociate cu Faza I a dezvoltării, însă pot fi efectuate în orice moment din procesul de dezvoltare a produsului medicinal.
Studiile ADME oferă, de asemenea, informații privind biodisponibilitatea – mai exact, fracțiunea din doza administrată de ingredient farmaceutic activ (IFA) care ajunge în sistemul circulator.
Studiile de biodisponibilitate (BD) și bioechivalență (BE)
Obiectivele studiului
Studiile de biodisponibilitate evaluează rata și gradul de absorbție a medicamentului. Acestea investighează concentrația în sânge, în timp, a ingredientului farmaceutic activ (IFA).
Medicamentele administrate pe căi diferite au profiluri de biodisponibilitate diferite. De exemplu, medicamentele administrate direct în sânge prin injecție intravenoasă (IV) au o biodisponibilitate de 100% imediat ce sunt administrate, în timp ce medicamentele administrate oral nu sunt biodisponibile decât după ce medicamentul este absorbit și intră în sistemul circulator.
Studiile de biodisponibilitate evaluează rata de absorbție a unui medicament măsurând concentrația maximă a medicamentului (Cmax) în sânge în momentul (Tmax) în care apare concentrația maximă. Aria de sub curbă (ASC) reprezintă expunerea totală a corpului la medicament și este utilizată pentru a studia gradul de absorbție.
Structura studiului
Aceste studii se desfășoară, în mod normal, în regim încrucișat și randomizat, folosind o doză unică administrată unor participanți sănătoși. Cercetătorii măsoară concentrația medicamentului și principalii metaboliți activi ai acestuia în sângele și plasma participanților.
Studiile de bioechivalență
Studiile de bioechivalență investighează relația între două formule diferite. Acestea studiază, de asemenea, rata și gradul de absorbție a medicamentului, însă le compară cu rata și gradul de absorbție a unui alt medicament sau a unei formule diferite a aceluiași medicament (medicamentul/formula de referință). Studiile de bioechivalență sunt utilizate pentru compararea medicamentelor generice cu cele de referință. Există criterii bine definite pe care un medicament trebuie să le îndeplinească înainte de a fi considerat bioechivalent cu un alt medicament.
Studiile privind efectul alimentelor
Obiectivele studiului
Studiile privind efectul alimentelor evaluează efectele alimentelor asupra ratei, gradului și biodisponibilității absorbției medicamentului administrat într-o anumită formulă. Informațiile privind studiile de efect al alimentelor sunt importante pentru instrucțiunile de administrare incluse în prospectul medicamentului (PM), care indică dacă medicamentul trebuie administrat pe stomacul gol sau în timpul meselor.
Structura studiului
Aceste studii sunt, în general, studii încrucișate cu doză unică, care compară două situați: participanți care nu au consumat niciun aliment și participanți care au consumat alimente bogate în grăsimi și calorii. Studiul are, în mod normal, două etape, este efectuat pe participanți sănătoși și utilizează cea mai mare concentrație anticipată de medicament.
Studiile privind insuficiența renală
Obiectivele studiului
Obiectivul studiilor privind insuficiența renală este de a evalua medicamentul la oameni cu diferite niveluri ale funcției renale (a rinichilor). Aceste studii colectează informații privind efectul excreției medicamentului din corp asupra funcției renale și urmăresc să obțină recomandări privind dozarea pentru pacienții cu insuficiență renală de diferite niveluri de gravitate.
Structura studiului
Studiile privind insuficiența renală sunt studii cu doză unică și grupuri paralele, efectuate pe voluntari sănătoși de ambele sexe (cca șase în fiecare grup). Grupurile sunt stratificate pe baza biomarkerilor funcției renale.
Studiile privind insuficiența hepatica
Obiectivele studiului
Obiectivul studiilor privind insuficiența hepatică este de a evalua medicamentul la oameni cu insuficiență hepatică (o suferință a ficatului) cu diferite niveluri de gravitate. Aceste studii examinează efectul farmacocineticii și metaboliților medicamentului asupra funcției hepatice și urmăresc să obțină recomandări privind dozarea pentru pacienții care suferă de insuficiență hepatică cu diferite niveluri de gravitate, din motive legate de eficacitate și/sau siguranță.
Structura studiului
Dacă rezultatele farmacocinetice ale studiilor anterioare sunt liniare și nu depind de timp, atunci studiile privind insuficiența hepatică iau, în general, forma unor studii cu grupuri paralele constând în voluntari sănătoși de ambele sexe (cca opt persoane), cu insuficiență hepatică de diferite niveluri. Grupurile de tratament sunt stratificate pe baza unor clasificări standard ale nivelurilor de insuficiență hepatică.
Dacă medicamentul este metabolizat de o enzimă care apare numai din cauza variațiilor genetice, participanții trebuie evaluați pe baza stării genotipurilor acestora.
Studiile de interacțiune a medicamentelor (interacțiuni între medicamente, IM)
Obiectivele studiului
Studiile de interacțiune a medicamentelor (numite studii IM) evaluează efectul medicației concomitente asupra farmacocineticii produsului medicamentos investigat (PMI), precum și efectul PMI asupra farmacocineticii altor medicamente administrate concomitent.
Structura studiului
Aceste studii sunt efectuate pe baza rezultatelor cercetărilor in vitro anterioare; este preferabil ca structura acestora să fie cea încrucișată. Studiile de interacțiune a medicamentelor sunt efectuate pe voluntari sănătoși sau pe pacienți dacă se dorește evaluarea obiectivelor finale farmacodinamice atunci când medicamentele sunt prea toxice (de exemplu, în cazul medicamentelor pentru cancer).
În mod normal, aceste studii utilizează structura încrucișată. Dozarea, intervalele de dozare, numărul de doze, căile de administrare și momentele de co-administrare trebuie concepute pentru maximizarea posibilității de detectare a unei interacțiuni și trebuie să simuleze cadrul clinic. Magnitudinea interacțiunii (inhibiției/inducției) este determinată pe baza modificării absorbției unuia dintre produsele medicamentoase investigate, calculată ca arie de sub curbă (ASC) pentru substanța respectivă.
Studiile QT aprofundate (QTA)
Obiectivele studiului
Intervalele QT reprezintă un mijloc de măsurare a ritmului cardiac. Un interval QT poate fi măsurat cu ajutorul unei electrocardiograme (ECG) și poate fi utilizat ca biomarker (imperfect) pentru evaluarea riscului ca un medicament să provoace aritmie. Pe ECG, activitatea electrică a inimii este măsurată și afișată sub forma unor unde cu etichetele „P”, „Q”, „R”, „S” și „T”. Intervalul QT este măsurat de la începutul undei Q la sfârșitul undei T.
Structura studiului
Studiile QTA sunt efectuate in vivo pentru toate entitățile moleculare noi (ENM). Acestea trebuie să aibă loc înainte de studiile din Faza III; indiferent de descoperirile studiilor in vitro sau ale celor non-clinice.
În mod normal, studiile QTA utilizează o singură doză, au structură încrucișată și sunt efectuate pe participanți sănătoși. Cercetătorii evaluează dozele terapeutice și supra-terapeutice (mai mari decât cele terapeutice) ale medicamentului comparându-l cu un control pozitiv (cum ar fi un antibiotic de largă utilizare, precum moxifloxacina) și un control negativ (placebo).
A2-5.03.4-V1.1