Last update: 19 listopada 2015


Wprowadzenie
Od wczesnych etapów procesu odkrywania i rozwoju leków dane gromadzone w badaniach nieklinicznych mają kluczowe znaczenie dla podejmowania decyzji dotyczących skuteczności i bezpieczeństwa — na przykład w przypadku planowania zarządzania ryzykiem i ograniczania ryzyka, warunków i specyfikacji związanych z dopuszczeniem do obrotu, stosowania leku dopuszczonego do obrotu oraz monitorowania bezpieczeństwa po wprowadzeniu na rynek (nadzór nad bezpieczeństwem farmakoterapii).
Informacje zgromadzone podczas badań nieklinicznych odgrywają główną rolę w podejmowaniu decyzji dotyczących:
- badań klinicznych,
- zarządzania ryzykiem oraz ograniczania ryzyka,
- wniosków o pozwolenie na dopuszczenie do obrotu,
- przepisywania leku pacjentowi,
- badań po wprowadzeniu produktu do obrotu,
- i w wielu innych sytuacjach.
Poniższy schemat przedstawia potrzeby i czynniki o kluczowym znaczeniu podczas procesu odkrywania i rozwoju leku. Informacje zgromadzone podczas badań nieklinicznych odgrywają kluczową rolę w określaniu tych potrzeb i czynników. W artykule przedstawiono ważną rolę badań nieklinicznych jako istotnego czynnika prognostycznego dla badań klinicznych z udziałem ludzi.
Główne czynniki wpływające na pomyślny rozwój leku — „Schemat 5 R”1
- Odpowiednie miejsce docelowe (ang. right target)
- Silna korelacja pomiędzy miejscem docelowym leku i chorobą
- Dostępne biomarkery o wartości prognostycznej
- Odpowiednia tkanka (ang. right tissue)
- Odpowiednia biodostępność i ekspozycja tkanki
- Definicja markerów farmakodynamicznych
- Przejrzyste informacje o przedklinicznej i klinicznej farmakokinetyce i farmakodynamice
- Wiedza o interakcjach z innymi lekami (interakcje leku z lekiem)
- Odpowiednie bezpieczeństwo (ang. right safety)
- Określony margines bezpieczeństwa
- Wiedza o drugorzędnych zagrożeniach farmakologicznych
- Wiedza o aktywnych metabolitach, genotoksyczności i interakcjach z innymi lekami
- Wiedza o niebezpiecznych działaniach ubocznych i innych problemach
- Odpowiedni pacjenci (ang. right patients)
- Identyfikacja populacji pacjentów najlepiej odpowiadających na leczenie
- Definicja relacji pomiędzy korzyścią a ryzykiem w danej populacji
- Odpowiedni potencjał komercyjny (ang. right commercial potential)
- Relacja kosztu do korzyści wobec przyszłych standardów opieki
- Koncentracja na dostępie do rynku
Od badań laboratoryjnych i na zwierzętach do pacjentów
Związku „kandydującego” nie można podać ludziom przed zebraniem wystarczającej ilości danych na temat jego profilu bezpieczeństwa i oczekiwanych efektów działania. Takie dane można uzyskać dzięki badaniom nieklinicznym, które dostarczają ważnych czynników prognostycznych, takich jak „potwierdzenie słuszności koncepcji”, proponowany schemat dawkowania, właściwe monitorowanie bezpieczeństwa i odpowiednie kryteria włączania i wykluczania.
Badania niekliniczne na komórkach (in vitro) i na zwierzętach (in vivo) powinny zatem:
- dowieść skuteczności związku „kandydującego”,
- dostarczyć informacji o profilu bezpieczeństwa związku „kandydującego” (na przykład dzięki badaniom maksymalnej tolerowanej dawki) oraz
- umożliwić oszacowanie działań związku „kandydującego”, których nie można badać z udziałem ludzi — na przykład wpływu związku na płód lub na kobietę ciężarną.
Ekstrapolacja wyników uzyskanych w badaniach na zwierzętach na ludzi
Ekstrapolacja danych zebranych podczas badań laboratoryjnych i badań na zwierzętach na zastosowanie leku u ludzi wymaga profesjonalnej oceny. Reguły przydatne w procesach ekstrapolacji opracowano i opisano w wytycznych Komitet ds. Produktów Leczniczych Stosowanych u Ludzi (CHMP)2 Europejskiej Agencji Leków (EMA) oraz Międzynarodowej Konferencji Harmonizacji (ICH)3. Wytyczne te określają rodzaje badań, które należy przeprowadzić przed rozpoczęciem badań klinicznych.
Problemy z programem badań nieklinicznych związku „kandydującego” mogą być przyczyną wnoszenia zastrzeżeń podczas oceny przez władze wniosku o pozwolenie na dopuszczenie do obrotu. Takie sytuacje prowadzą do pytań o przydatność modeli nieklinicznych zastosowanych w przypadku proponowanych wskazań, w których związek „kandydujący” ma być stosowany do leczenia ludzi. Aby uniknąć takich problemów, należy starannie zaplanować badania niekliniczne, tak aby oczekiwania wynikające z badań laboratoryjnych i na zwierzętach mogły stanowić zadowalające czynniki prognostyczne.
Zakres i możliwości programu rozwoju nieklinicznego, które muszą zostać pozytywnie zakończone przed rozpoczęciem badań klinicznych różni się w zależności od następujących czynników:
- rodzaju i stopnia nasilenia choroby docelowej,
- rozmiaru i dynamiki populacji, do której leczenia ma służyć związek „kandydujący”,
- fazy badania klinicznego (Faza I, II, III i Faza IV po wprowadzeniu produktu do obrotu) oraz
- przewidywanej dawki i czasu trwania leczenia u ludzi.
Powyższe czynniki są stosowane do określenia rodzajów testów lub modeli zwierzęcych stosowanych w programie badań nieklinicznych.
Wiele firm prosi odnośne władze (na przykład Europejską Agencję Leków lub władze krajowe) o porady naukowe dotyczące badań nieklinicznych. Porady naukowe pozwalają firmie zapewnić przeprowadzenie odpowiednich testów i badań, tak aby podczas oceny wniosku o pozwolenie na dopuszczenie do obrotu nie pojawiły się poważne wątpliwości co do planu badań. Kierowanie do takich agencji próśb o rady i postępowanie zgodnie z nimi zwiększa prawdopodobieństwo uzyskania pozytywnego wyniku na etapie wnioskowania o pozwolenie na dopuszczenie do obrotu. Porady są udzielane zgodnie z aktualną wiedzą naukową i są oparte na dokumentacji dostarczonej przez firmę.
Dane z badań nieklinicznych są najważniejsze na wczesnych etapach procesu rozwoju związku „kandydującego” (zob. Ryc. 1). Do czasu, kiedy będzie możliwe przepisywanie leku (po uzyskaniu zezwolenia na dopuszczenie do obrotu), wiele danych nieklinicznych dotyczących bezpieczeństwa i skuteczności leku zostanie zastąpionych danymi z badań z udziałem ludzi. Jednak w niektórych wypadkach — na przykład w odniesieniu do oddziaływania związku „kandydującego” na rozwój chorób nowotworowych lub na zdolności reprodukcyjne — względy etyczne nie pozwalają na gromadzenie danych w ramach badań z udziałem ludzi. W takich wypadkach stosowanie kliniczne nowych leków będzie odbywać się przez dłuższy czas na podstawie danych nieklinicznych. Ostatecznie jednak te dane także będą musiały zostać zastąpione przez dane zebrane podczas leczenia ludzi, na przykład w ramach zarządzania cyklem życia po wprowadzeniu produktu na rynek i nadzoru nad bezpieczeństwem farmakoterapii.
-
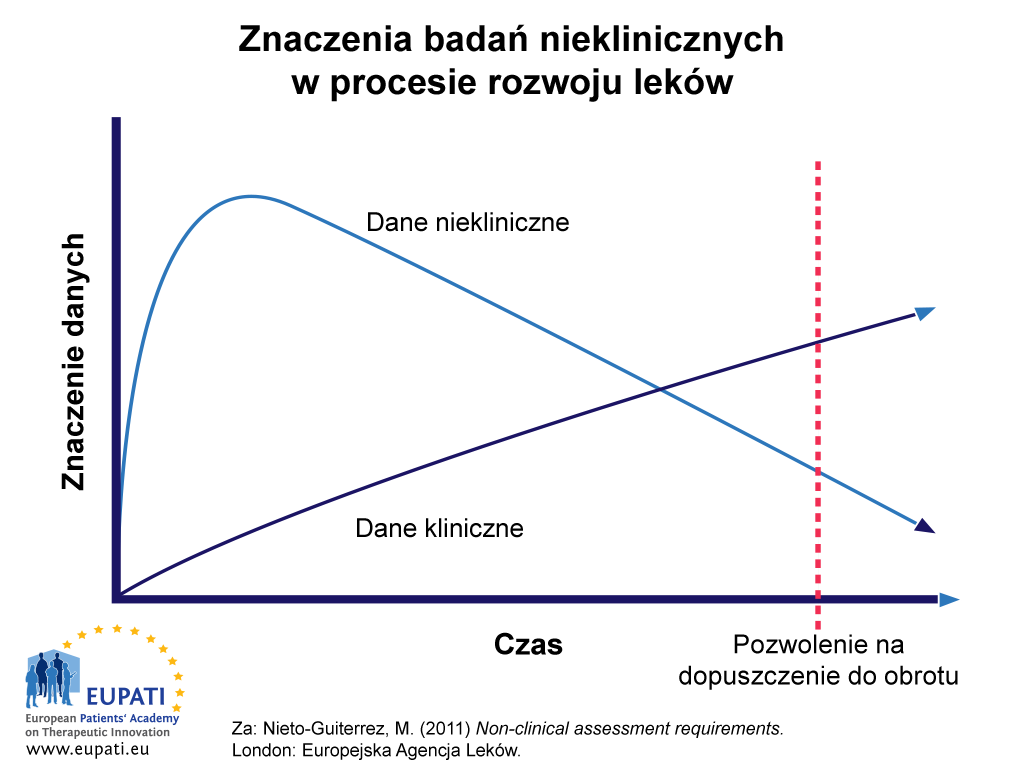
- Dane niekliniczne mają znacznie większe znaczenie na wczesnym etapie procesu rozwoju leku, ale w przebiegu czasu ich znaczenie zostaje przewyższone przez dane kliniczne.
Rycina 1 przedstawia względną wagę i znaczenie danych z badań nieklinicznych w procesie rozwoju leków w zależności od czasu. Do późniejszego okresu rozwoju leku dane z badań nieklinicznych mają większe znaczenie niż dane kliniczne.
Idealną sytuacją jest, jeśli wszystkie zastrzeżenia dotyczące bezpieczeństwa stwierdzone w trakcie badań nieklinicznych zostaną wyjaśnione do czasu złożenia wniosku o pozwolenie na dopuszczenie do obrotu. Jednak w czasie składania i oceny dossier ważne problemy dotyczące bezpieczeństwa mogą nadal pozostawać niewyjaśnione, na przykład rakotwórczość, genotoksyczność, domieszki genotoksyczne, toksyczność reprodukcyjna i hepatotoksyczność.
Zagadnienia etyczne
Akceptowalność wykorzystywania zwierząt jako modeli do oceny ryzyka u ludzi i stosowanie takich modeli do naśladowania chorób występujących u ludzi została określona w Deklaracji Helsińskiej.4 Deklaracja Helsińska przedstawia etyczne i naukowe uzasadnienie dla pierwszego zastosowania związku „kandydującego” u zdrowych ochotników. Co więcej, Deklaracja stanowi, że badania biomedyczne powinny opierać się na właściwie przedstawionych badaniach laboratoryjnych i badaniach na zwierzętach oraz na dokładnej znajomości literatury naukowej. Konieczne jest zapewnienie dobrostanu zwierząt wykorzystywanych do badań.
Badania niekliniczne: Czynniki prognostyczne odpowiednie do badań z udziałem ludzi?
W przeszłości problemy dotyczące wartości prognostycznej badań nieklinicznych były związane z farmakokinetyką, farmakodynamiką (skutecznością) i niemożliwymi do przewidzenia na etapie badań nieklinicznych aspektami bezpieczeństwa u ludzi. Wiele nowych technologii in silico (modele komputerowe), farmakogenomika, biomarkery i nowatorskie schematy badań klinicznych rozwija się szybko i pozytywnie wpływa na wartość prognostyczną badań nieklinicznych.
Inne materiały
- Deklaracja Helsińska jest dostępna w językach angielskim, hiszpańskim i francuskim na stronie Retrieved 13 July, 2021, from https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (pobrano 23 września 2015). Jest także dostępna w językach niemieckim, japońskim, portugalskim, czeskim i węgierskim na stronie http://www.wma.net/en/20activities/10ethics/10helsinki/ (pobrano 23 września 2015)
Piśmiennictwo
- Adapted from Cook, D., Brown, D., Alexander, R., March, R., Morgan, P., Satterthwaite, G., & Pangalos, M. (2014). Lessons learned from the fate of AstraZeneca's drug pipeline: A five-dimensional framework. Nature Reviews Drug Discovery, 13, 419-431. doi:10.1038/nrd4309
- European Medicines Agency. (2015) Non-clinical guidelines. Retrieved 24 July, 2015, from http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000083.jsp&mid=WC0b01ac0580027548
- International Conference on Harmonisation (2015). ICH Guidelines. Retrieved 24 July, 2015, from http://www.ich.org/products/guidelines.html
- World Medical Association. (2013) WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects. Retrieved 24 July, 2015, from http://www.wma.net/en/30publications/10policies/b3/
- Nieto-Guiterrez, M. (2011) Non-clinical Assessment Requirements. Brussels: European Medicines Agency. Retrieved 24 July, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2011/06/WC500107868.pdf
Załączniki
- Prezentacja: Rozwój niekliniczny
Size: 396,742 bytes, Format: .pptx
Prezentacja dotycząca nieklinicznego opracowania leków. W tej prezentacji przedstawiono cele rozwoju nieklinicznego, działania na zapleczu (w tym produkcję niezbędnej substancji czynnej), rodzaje badań nieklinicznych, specyfikę modeli zwierzęcych, uwagi dotyczące terminów i czasu trwania oraz wyniki badań nieklinicznych mogące przerwać prace nad substancją „kandydującą”. - Prezentacja: Rozwój niekliniczny
Size: 396,742 bytes, Format: .pptx
Prezentacja dotycząca nieklinicznego opracowania leków. W tej prezentacji przedstawiono cele rozwoju nieklinicznego, działania na zapleczu (w tym produkcję niezbędnej substancji czynnej), rodzaje badań nieklinicznych, specyfikę modeli zwierzęcych, uwagi dotyczące terminów i czasu trwania oraz wyniki badań nieklinicznych mogące przerwać prace nad substancją „kandydującą”.
A2-2.02.1-v1.3