

Introduction
From early on in the medicines discovery and development process, data gathered in non-clinical studies is crucial for efficacy and safety decision making – for instance, regarding risk-management planning, risk mitigation, the conditions and specifications of market authorisation, the use of the medicine on the market, and post-marketing safety monitoring (pharmacovigilance).
The information gathered during non-clinical studies plays a key role in decisions:
- on clinical trials,
- on risk management and mitigation,
- on marketing authorisation applications,
- on the prescription of a medicine to a patient,
- on post-marketing or monitoring studies,
- and more.
The following framework illustrates needs and factors that are crucial during the medicines discovery and development process. Non-clinical information plays a key role in determining these needs and factors. This article explores the important role of non-clinical studies as an important predictor for clinical studies in human patients.
The keys to successful medicines development – the ‘Five R Framework’1
- Right target
- Strong correlation between medicine’s target and the disease
- Available and predictive biomarkers
- Right tissue
- Adequate bioavailability and tissue exposure
- Definition of pharmacodynamic biomarkers
- Clear understanding of pre-clinical and clinical pharmacokinetics and pharmacodynamics
- Understanding of interactions with other medicines (drug-drug interactions)
- Right safety
- Clear safety margins
- Understanding of secondary pharmacology risks
- Understanding of reactive metabolites, genotoxicity, and interactions with other medicines
- Understanding of dangerous side effects and other liabilities
- Right patients
- Identification of the most responsive patient population
- Definition of benefit-risk relationship for the given population
- Right commercial potential
- Cost-benefit versus future standard of care
- Focus on market access
From laboratory and animal studies to patients
A candidate compound cannot be administered to human patients before enough supporting information has been gathered regarding its safety profile and the effects it is expected to have. Non-clinical studies provide this supporting information by providing important predictors, such as ‘proof of concept’, a proposed dosage regimen, adequate safety monitoring, and appropriate inclusion and exclusion criteria.
Non-clinical cell (in vitro) and animal studies (in vivo) should therefore:
- demonstrate the efficacy of the candidate compound,
- provide knowledge on the safety profile of the candidate compound, for instance studies investigating maximum tolerated dose, and
- estimate the effects of the candidate compound that cannot be studied in humans – for instances, the compound’s effect on foetuses or in pregnant women.
Extrapolating from animals to humans
Extrapolating from information gathered in laboratory and animal studies to human application of a medicine requires professional judgement. Useful rules for the extrapolation processes have been developed and described in the guidelines of the Committee for Human Medicinal Products (CHMP)2 of the European Medicines Agency (EMA) and the International Conference on Harmonisation (ICH)3. These guidelines specify the types of studies that must take place before clinical trials can be conducted.
Issues with the non-clinical programme of a candidate compound can be the cause for objections during the evaluation of the marketing authorisation application (MAA) during regulatory review. Such cases lead to questions about the relevance of the non-clinical models used for the proposed indication that the candidate compound is intended to treat in humans. In order to avoid these issues, non-clinical studies must be carefully planned so that the expectations generated by laboratory and animal studies can function as satisfactory predictors.
The extent and scope of the non-clinical programme that must be satisfactorily completed before clinical trials may be initiated varies according to the following factors:
- the type and severity of the targeted disease,
- the population size and dynamics that the candidate compound is intended to treat,
- the clinical trial phase (Phase I, II, III, post-marketing Phase IV), and
- the anticipated dose and duration of the treatment in humans.
These considerations are used to justify the kinds of tests or animal models used during the non-clinical programme.
Many companies seek scientific advice on non-clinical studies from regulatory authorities (for instance, the European Medicines Agency (EMA) or national competent authorities). This scientific advice helps the company make sure that the appropriate tests and studies are conducted, so that no major objections regarding the design of the tests are likely to be raised during the MAA. Seeking and following these agencies’ advice increases the probability of a positive outcome at the MAA stage. The advice is given in the light of the current scientific knowledge, and is based on the documentation provided by the company.
Non-clinical data is most important in early parts of the development process of a candidate compound (see Figure 1). By the time a medicine is available for prescription (post-MAA), much of the non-clinical data on safety and efficacy will be replaced by data from clinical trials in human patients. However, in some cases – for instance the effect of a candidate compound on cancer development or in reproduction – ethical considerations prevent the gathering of data in human subjects. In these cases, the clinical use of new medicines will be directed by non-clinical data for a longer time. Eventually, however, this too will need to be replaced by data gathered on these instances, for instance as part of post-marketing life-cycle management and pharmacovigilance.
-
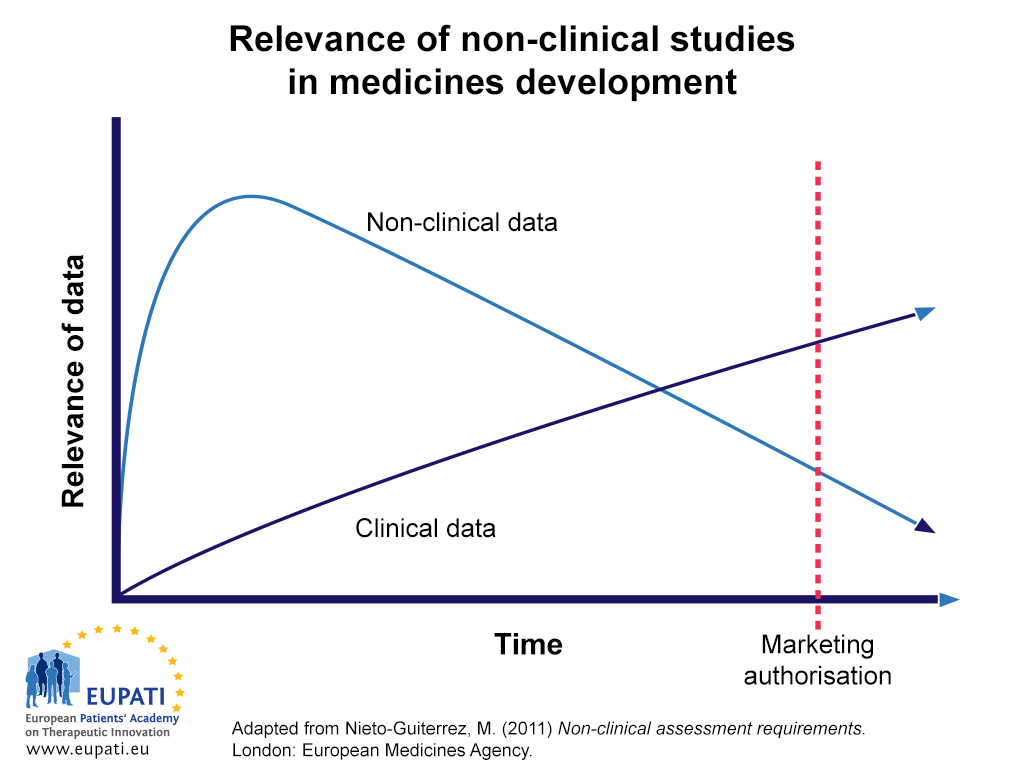
- While non-clinical data is much more relevant to the medicines development process early on, over time its relevance is eclipsed by that of clinical data.
Figure 1 demonstrates the relative importance of and reliance on non-clinical data in the medicines development process over time. Data from non-clinical studies are relied on more than clinical data until later in the development process.
Ideally, all non-clinical safety concerns raised during the development period should be solved by the time of the MAA. However, at the time of the dossier submission and assessment, major causes for safety concern may still remain, including for instance carcinogenicity, genotoxicity, genotoxic impurities, reproductive toxicity, and hepatotoxicity.
Ethical considerations
The acceptability of using animals as models of risk assessment for human and for the use of these models to mimic human diseases is laid out by the Declaration of Helsinki.4 The Declaration of Helsinki provides the ethical and scientific justification for the first exposure of candidate compounds to healthy volunteers. Furthermore, the Declaration states that biomedical research should be based on adequately performed laboratory and animal experimentation and on thorough knowledge of the scientific literature. The welfare of animals used for research must be respected.
Non-clinical studies: Appropriate predictors for studies in humans?
Historically, challenges to the predictive value of non-clinical studies have been related to pharmacokinetics, pharmacodynamics (efficacy), and safety aspects in humans that cannot easily be predicted by non-clinical studies. Many new technologies in silico (computer models), pharmacogenomics, biomarkers, and novel exploratory designs for clinical trials are in rapid development, and all have a positive influence on the predictive value of non-clinical studies.
Further Resources
- The Declaration of Helsinki is available in English, Spanish, and French at https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (Retrieved 4, July 2021). It is also available in Czech, German and Portuguese, at https://web.archive.org/web/20160517043747/http://www.wma.net/en/20activities/10ethics/10helsinki (Retrieved 4 July 2021)
References
- Adapted from Cook, D., Brown, D., Alexander, R., March, R., Morgan, P., Satterthwaite, G., & Pangalos, M. (2014). Lessons learned from the fate of AstraZeneca's drug pipeline: A five-dimensional framework. Nature Reviews Drug Discovery, 13, 419-431. doi:10.1038/nrd4309
- European Medicines Agency. (2015) Non-clinical guidelines. Retrieved 24 July, 2015, from http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000083.jsp&mid=WC0b01ac0580027548
- International Conference on Harmonisation (2015). ICH Guidelines. Retrieved 24 July, 2015, from http://www.ich.org/products/guidelines.html
- World Medical Association. (2013) WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects. Retrieved 4 July, 2021, from https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/
- Nieto-Guiterrez, M. (2011) Non-clinical Assessment Requirements. Brussels: European Medicines Agency. Retrieved 24 July, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2011/06/WC500107868.pdf
Attachments
- Presentation: Non-Clinical Development
Size: 478,517 bytes, Format: .pptx
Presentation on aspects of non-clinical development, including its aims, background activities, and the different types of non-clinical study.
A2-2.02.1-v1.3
- Presentation: Non-Clinical Development