

Wprowadzenie
Aby lek mógł zostać wprowadzony zgodnie z prawem na rynek w Europejskim Obszarze Gospodarczym (EOG), musi uzyskać pozwolenie na dopuszczenie do obrotu.1 To pozwolenie ma przede wszystkim zagwarantować, że bezpieczne, skuteczne i wysokiej jakości leki będą szybko dostępne dla mieszkańców EOG. Należy zaznaczyć, że procedury umożliwiające wprowadzenie na rynek urządzeń medycznych różnią się od tych, którym podlegają inne produkty lecznicze.
Odnośne władze krajowe (lub w skrócie odnośne władze) — są to krajowe agencje administracji państwowej odpowiedzialne za ocenę wniosków o wydanie pozwolenia na dopuszczenie do obrotu (MAA) i przyznające pozwolenia na dopuszczenie do obrotu produktów leczniczych, które są wprowadzane na ich rynki z zastosowaniem procedur krajowych, zdecentralizowanych lub wzajemnego uznania. Europejska Agencja Leków (EMA) w Londynie odpowiada za koordynację naukowej oceny wniosków o pozwolenie na dopuszczenie do obrotu w Europie produktów leczniczych podlegających procedurze centralnej. Ocenę przeprowadzają eksperci władz krajowych. Po pozytywnej weryfikacji Komisja Europejska wydaje pozwolenie na dopuszczenie do obrotu.
Procedury związane z uzyskaniem pozwolenia na dopuszczenie do obrotu
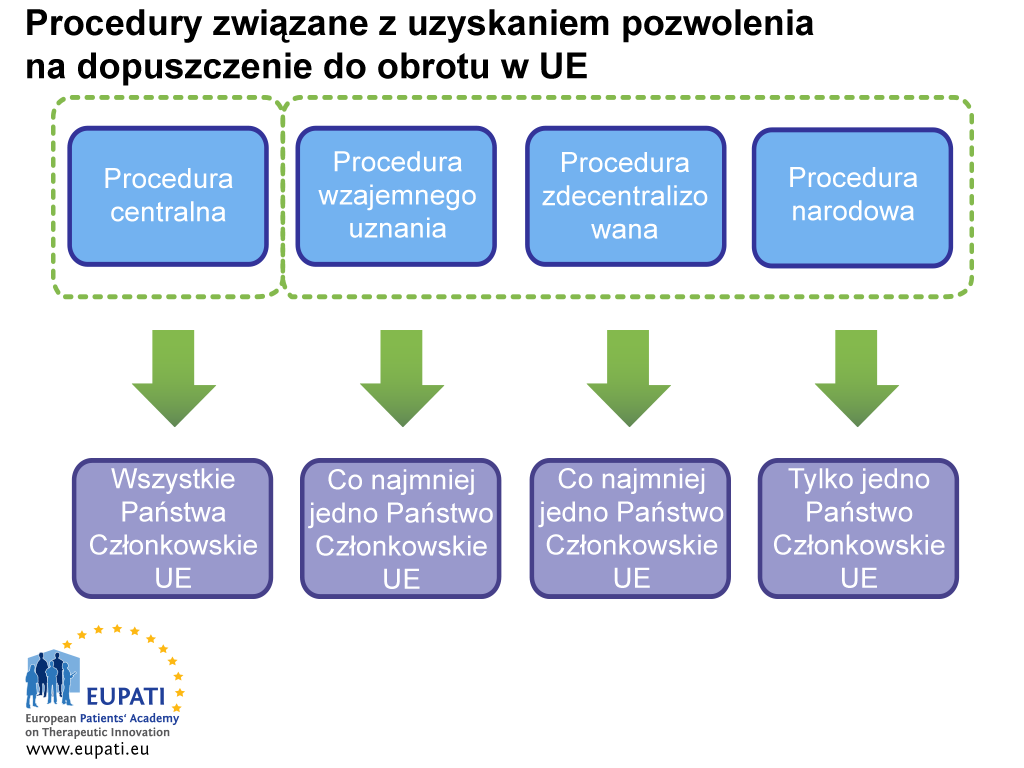
Pozwolenie na dopuszczenie do obrotu można uzyskać dwiema drogami: za pośrednictwem procedury centralnej lub niecentralnej. Ta ostatnia obejmuje procedurę zdecentralizowaną, procedurę wzajemnego uznania i procedurę narodową. W każdym systemie władze (EMA lub odnośne władze krajowe) i podmioty odpowiedzialne posiadające pozwolenia na dopuszczenie do obrotu podlegają właściwym dla tego systemu przepisom i realizują odpowiednie obowiązki.
Procedura centralna (CP)
Większość nowych leków korzysta z tej drogi. Firma przesyła jeden wniosek do Europejskiej Agencji Leków. Komitet ds. produktów leczniczych stosowanych u ludzi jest częścią Europejskiej Agencji Leków, (CHMP). W jego skład wchodzi jeden członek z każdego Państwa Członkowskiego UE i kraju EOG oraz pięciu ekspertów naukowych, kieruje on do Komisji Europejskiej rekomendację dotyczącą rejestracji (lub nie) leku. Jeśli lek zostanie zarejestrowany tą drogą, pozwolenie na dopuszczenie do obrotu zostaje przyznane wszystkim krajom UE i EOG. Procedura oceny naukowej może trwać maksymalnie 210 dni roboczych, ale ten czas można wstrzymać (tzw. „zatrzymanie czasu”).
Procedura zdecentralizowana i procedura wzajemnego uznania
W przypadku tych procedur pozwolenie na dopuszczenie do obrotu otrzymuje jednocześnie kilka Państw Członkowskich, a jedno z nich obejmuje rolę lidera jako Państwo Referencyjne (ang. Reference Member State, RMS). Jeśli procedura zostanie zakończona pomyślnie, lek uzyskuje pozwolenie na dopuszczenie do obrotu w Państwie Referencyjnym i w Zainteresowanych Państwach Członkowskich (ang. Concerned Member States, CMS). Procedury te opierają się na zasadzie uznania przez Zainteresowane Państwa Członkowskie oceny sformułowanej przez Państwo Referencyjne. Większość leków generycznych jest rejestrowana za pośrednictwem procedury zdecentralizowanej.
Procedura narodowa
Niezależne procedury narodowe są ściśle ograniczone do leków, które mają być zarejestrowane i dopuszczone do obrotu tylko w jednym Państwie Członkowskim. Procedura jest obecnie rzadko stosowana w przypadku nowych produktów.
Wybór procedury uzyskania pozwolenia na dopuszczenie do obrotu
Zwykle firma farmaceutyczna może samodzielnie wybierać procedurę do zastosowania. Jest to decyzja biznesowa, a na wybór danej procedury mogą wpływać różne przyczyny. Procedura centralna obowiązuje w przypadku:
- produktów uzyskanych za pomocą procesów biotechnologicznych,
- leków do terapii zaawansowanej (na przykład terapii genowej, terapii komórkami somatycznymi lub leków uzyskanych w wyniku inżynierii tkankowej),
- sierocych produktów leczniczych,
- leków przeznaczonych do leczenia zakażenia wirusem HIV lub AIDS, raka, chorób neurodegeneracyjnych, autoimmunologicznych i innych dysfunkcji układu immunologicznego, chorób wirusowych i cukrzycy).
Procedurę centralną można także stosować w odniesieniu do leków stanowiących istotną innowację terapeutyczną, naukową lub techniczną, lub będących przedmiotem zainteresowania zdrowia publicznego (np. leki wspomagające utratę masy ciała).
Jeśli firma posiada pozwolenie na dopuszczenie danego leku do obrotu, ale chce rozwijać go tak, aby można było go stosować z innych wskazań, zwykle musi złożyć wniosek o całkowicie nowe pozwolenie na dopuszczenie do obrotu.
-
- W uzyskanie pozwolenia na dopuszczenie leku do obrotu zaangażowane są różne strony zależnie od procedury wybranej (lub obowiązującej) sponsora.
Ocena naukowa leków
Niezależnie od procedury, w przypadku każdego leku ocenianego pod kątem możliwości dopuszczenia do obrotu należy sporządzić standardowe „dossier”. Wspólny Dokument Techniczny (CTD) jest to międzynarodowo uznany zharmonizowany format obowiązujący w przypadku wniosków przesyłanych do odnośnych władz. Wspólny Dokument Techniczny to zestaw dokumentów, w których wnioskodawca musi dowieść, że jakość danego produktu medycznego jest zgodna z wymaganiami oraz że jego stosowanie jest bezpieczne i skuteczne.
Odmowa wydania pozwolenia na dopuszczenie do obrotu
Odmowa wydania pozwolenia na dopuszczenie do obrotu ma miejsce w następujących sytuacjach:
- Stosunek korzyści do ryzyka w przypadku danego produktu leczniczego nie zostanie uznany za dobry lub
- Skuteczność terapeutyczna nie została wystarczająco uzasadniona przez wnioskodawcę lub
- Skład ilościowy i jakościowy są różne od zadeklarowanych.
Ważność pozwolenia na dopuszczenie do obrotu i działania kontrolne
Pozwolenie na dopuszczenie do obrotu jest przedłużane po pięciu latach przez odnośne władze Państwa Członkowskiego, w którym dokonano rejestracji, na podstawie ponownej oceny stosunku korzyści i ryzyka. Po przedłużeniu, pozwolenie na dopuszczenie do obrotu jest ważne przez nieograniczony czas, chyba że odnośne władze uznają, na podstawie uzasadnionych informacji dotyczących nadzoru nad bezpieczeństwem farmakoterapii, że należy wyznaczyć kolejny pięcioletni okres, po którym będzie wymagane przedłużenie.
Wszystkie podmioty odpowiedzialne posiadające pozwolenie na dopuszczenie do obrotu są zobowiązane do obserwowania danych o bezpieczeństwie stosowania i przestrzegania innych wymagań dotyczących okresu po dopuszczeniu produktu do obrotu. Po wprowadzeniu na rynek leki muszą być nadal monitorowane (nadzór nad bezpieczeństwem farmakoterapii), aby zapewnić, że wszelkie kwestie mogącą wpłynąć na profil bezpieczeństwa leku zostały wykryte, zbadane, a wszystkie konieczne działania zostały podjęte.
W czasie, gdy lek znajduje się w obrocie, mogą stać się dostępne nowe dane lub informacje dotyczące jego skuteczności, bezpieczeństwa stosowania lub produkcji. W takim wypadku obowiązkiem podmiotu odpowiedzialnego jest przedłożenie odnośnym władzom wniosku o zmianę w pozwoleniu na dopuszczenie do obrotu we wszystkich Państwach Członkowskich, w których poprzednio zarejestrowano ten produkt leczniczy.
Zaangażowanie pacjentów w związku z pozwoleniem na dopuszczenie do obrotu
Zaangażowanie pacjentów w działania związane z udzieleniem pozwolenia na dopuszczenie do obrotu jest tradycyjnie bardzo niewielkie. Jednak niektóre organy regulacyjne zastanawiają się nad możliwością włączenia pacjentów w procedury uzyskiwania pozwolenia na dopuszczenie do obrotu — na przykład za pomocą konsultacji z pacjentami i/lub obecności przedstawicieli pacjentów w Komisji ds. leków stosowanych u ludzi w Agencji ds. Regulacji Leków i Produktów Ochrony Zdrowia w Wielkiej Brytanii. Pacjenci zostali już uwzględnieni w dyskusjach na temat stosunku korzyści i ryzyka w Europejskiej Agencji Leków jako Członkowie Komitetu ds. nadzoru nad bezpieczeństwem farmakoterapii, poprzez udział w procedurach dotyczących uzyskiwania opinii naukowych/pomocy w kwestiach regulacyjnych oraz w spotkaniach naukowych grup doradczych. Istnieje także pilotażowa inicjatywa umożliwiająca pacjentom udział w pracach Komitetu ds. Produktów Leczniczych Stosowanych u Ludzi na etapie ustnych wyjaśnień poprzedzających podjęcie decyzji.
Organizacje pacjentów mogą także wpływać na prawodawstwo krajowe/UE w kwestiach związanych z pozwoleniem na dopuszczenie do obrotu.
Piśmiennictwo
- The European Economic Area (EEA) unites the EU Member States and the three EEA EFTA States (Iceland, Liechtenstein, and Norway) into the internal market governed by the same basic rules. These rules aim to enable goods, services, capital, and persons to move freely about the EEA in an open and competitive environment, a concept referred to as the four freedoms.
Załączniki
- Pozwolenie na dopuszczenie do obrotu
Size: 766,633 bytes, Format: .pptx
Więcej informacji na temat procedur związanych z uzyskiwaniem pozwolenia na dopuszczenie do obrotu produktu leczniczego w Europejskim Obszarze Gospodarczym (EOG).
A2-5.07-v1.1