

Introduction
All medicines must have a Marketing Authorisation (MA) in order to be put on the market legally in the European Economic Area (EEA).1 The ultimate purpose of marketing authorisation is to ensure that safe, effective, and high-quality medicines can quickly be made available to citizens across the EEA. It is important to mention that medical devices must follow different processes than other medicinal products before they can be made available on the market.
National Competent Authorities (NCAs) – in short, ‘regulatory authorities’ – are national government agencies that are responsible for the evaluation of Marketing Authorisation Applications (MAAs) and granting MAs for medicinal products that are placed on their markets via national, decentralised, or mutual recognition procedures. The European Medicines Agency (EMA) in London is responsible for the coordination of the scientific evaluation of applications for European MAs for medicinal products under the Centralised Procedure (CP). The assessment is performed by NCA experts. After positive evaluation, the European Commission issues the MA.
Marketing authorisation procedures
There are two ways to obtain an MA: via the Centralised Procedure (CP) or via a non-centralised procedure. The latter includes the Decentralised Procedure (DCP), the Mutual Recognition Procedure (MRP), and the National Procedure. Each system has its own legal provisions and responsibilities for the competent authority (EMA or NCAs) and MA holders.
The Centralised Procedure (CP)
Most new medicines take this route. Industry submits a single application to the EMA. The EMA Committee for Human Medicinal Products (CHMP) – which consists of one member appointed by each of the EU Member States and EEA countries and five expert scientists – recommends to the European Commission if a medicine should be approved. If a medicine is approved in this way, a license for marketing the medicine is granted for all EU and EEA countries. This scientific evaluation process takes up to a maximum of 210 active days, but this count can be paused (called a ‘clock stop’).
Decentralised Procedure (DCP) and Mutual Recognition Procedure (MRP)
In these procedures, marketing authorisations are applied for in several Member States at the same time, with one country taking the lead as the Reference Member State (RMS). If successful, the medicine is approved for marketing in the RMS and the other involved countries (Concerned Member States, CMSs). These procedures are based on the principle of recognition of the RMS’s assessment by the CMSs. Most generic medicines are approved through the DCP.
National Procedure
Independent national procedures are strictly limited to medicines which are to be authorised and marketed in only one Member State (MS). This procedure is nowadays rarely followed for new products.
Selecting a marketing authorisation procedure
A pharmaceutical company is normally free to decide which procedure to use. This is a business decision that may include a variety of reasons to opt for a specific procedure. The CP is compulsory for:
- products derived from biotechnology processes,
- advanced-therapy medicines (such as gene-therapy, somatic cell therapy, or tissue-engineered medicines)
- orphan medicinal products,
- medicines intended for the treatment of HIV or AIDS, cancer, neurodegenerative disorders, auto-immune and other immune dysfunction, viral diseases, or diabetes.
The CP can also be used for medicines that are a significant therapeutic, scientific, or technical innovation or in the interest of public health, such as weight-loss medicine.
If a company wants to develop an existing medicine with a marketing authorisation for a different indication, then usually they must apply for a completely new marketing authorisation.
-
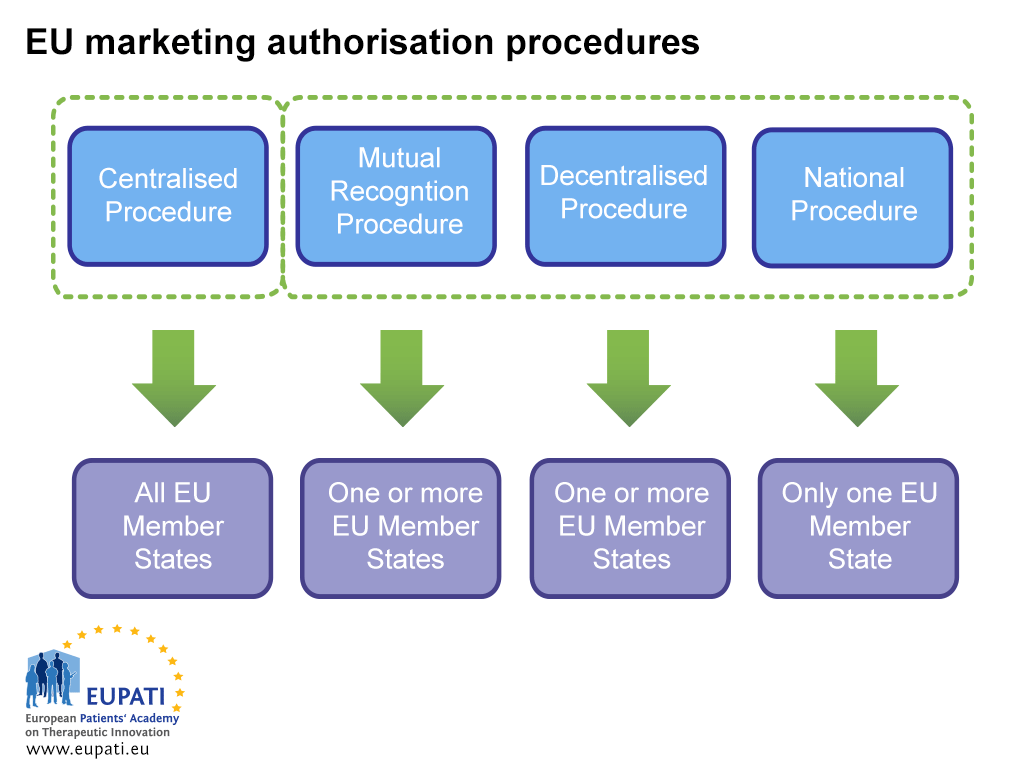
- Different actors are involved in the marketing authorisation of a medicine depending on which procedure the sponsor opts (or is obliged) to follow.
Scientific assessment of medicines
Regardless of the procedure, each medicine needs a standard ‘dossier’ for marketing authorisation assessment. The Common Technical Document (CTD) is an internationally recognised harmonised format that must be followed for applications intended to be submitted to regulatory authorities. The CTD consists of a set of documents in which the applicant must demonstrate that the medicinal product has the required quality and that it is safe and efficacious.
Refusal of a marketing authorisation
An MA will be refused, in cases where the medicinal product:
- Benefit-risk balance is not considered to be favourable, or
- Therapeutic efficacy is insufficiently substantiated by the applicant, or
- Qualitative and quantitative composition is not as declared.
Validity of marketing authorisation and follow up
The MA is renewed after five years on the basis of a re-evaluation of the benefit-risk balance by the Competent Authority of the authorising Member State. Once renewed, the MA is valid for an unlimited period, unless the Competent Authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal.
All marketing authorisation holders (MAHs) are required to provide follow up safety data and to follow other post-marketing requirements. Once placed on the market, medicines must in fact continue to be monitored (pharmacovigilance) to assure that any aspect that could impact the safety profile of a medicine is detected and assessed and that necessary measures are taken.
Whilst on the market, new data or information on the medicine’s efficacy, safety, or production may become available. In this case, it is the responsibility of the MAH to send a ‘variation’ application of a valid marketing authorisation to the Competent Authority(ies) in all the MSs that have previously authorised the medicinal product.
Patients’ involvement in marketing authorisation
Patients’ involvement in marketing authorisation has traditionally been very limited. Some competent authorities are, however, thinking about how to possibly involve patients in marketing authorisation procedures – for instance, through patient consultation and/or representation in the Commission on Human Medicines within the Medicines and Healthcare products Regulatory Agency (MHRA) in the United Kingdom. Patients are already involved in benefit-risk discussions at the EMA as members of the Pharmacovigilance Committee (PRAC), through their participation in scientific advice/protocol assistance procedures, and in Scientific Advisory Group (SAG) meetings. There is also a pilot initiative involving patients in the CHMP during oral explanations preceding decision-making.
Patient advocates also have the opportunity to influence national/EU legislation concerning marketing authorisation.
References
- The European Economic Area (EEA) unites the EU Member States and the three EEA EFTA States (Iceland, Liechtenstein, and Norway) into the internal market governed by the same basic rules. These rules aim to enable goods, services, capital, and persons to move freely about the EEA in an open and competitive environment, a concept referred to as the four freedoms.
Attachments
- Presentation: Marketing Authorisation
Size: 444,038 bytes, Format: .pptx
Find out more about the procedures involved in obtaining a marketing authorisation for a medicinal product in the European Economic Area.
A2-5.07-v1.1