Last update: 30 septembre 2015


Introduction
Tous les médicaments doivent avoir une autorisation de mise sur le marché (AMM) afin d’être commercialisés légalement dans l’Espace économique européen (EEE).1 L’objectif ultime de cette autorisation est d’assurer la disponibilité rapide de médicaments sûrs, efficaces et de haute qualité pour les citoyens de l’EEE. Il est important de mentionner que les instruments médicaux doivent suivre un parcours différent de celui des produits pharmaceutiques avant de pouvoir être commercialisés.
Les autorités nationales compétentes (NCA), ou « autorités réglementaires », sont des organismes gouvernementaux à l’échelle nationale responsables d’évaluer les demandes d’autorisation de mise sur le marché (DAMM) et d’octroyer des AMM pour les produits pharmaceutiques placés sur leur marché par le biais de procédures nationales, décentralisées ou de reconnaissance mutuelle. L’Agence européenne des médicaments (EMA) se trouve à Londres. Elle est responsable de la coordination de l’évaluation scientifique des demandes d’AMM européennes pour les produits pharmaceutiques dans le cadre de la procédure centralisée (CP). Cette évaluation est menée par des experts d’ANC Suite aux résultats positifs de l’évaluation, la Commission européenne accorde l’AMM.
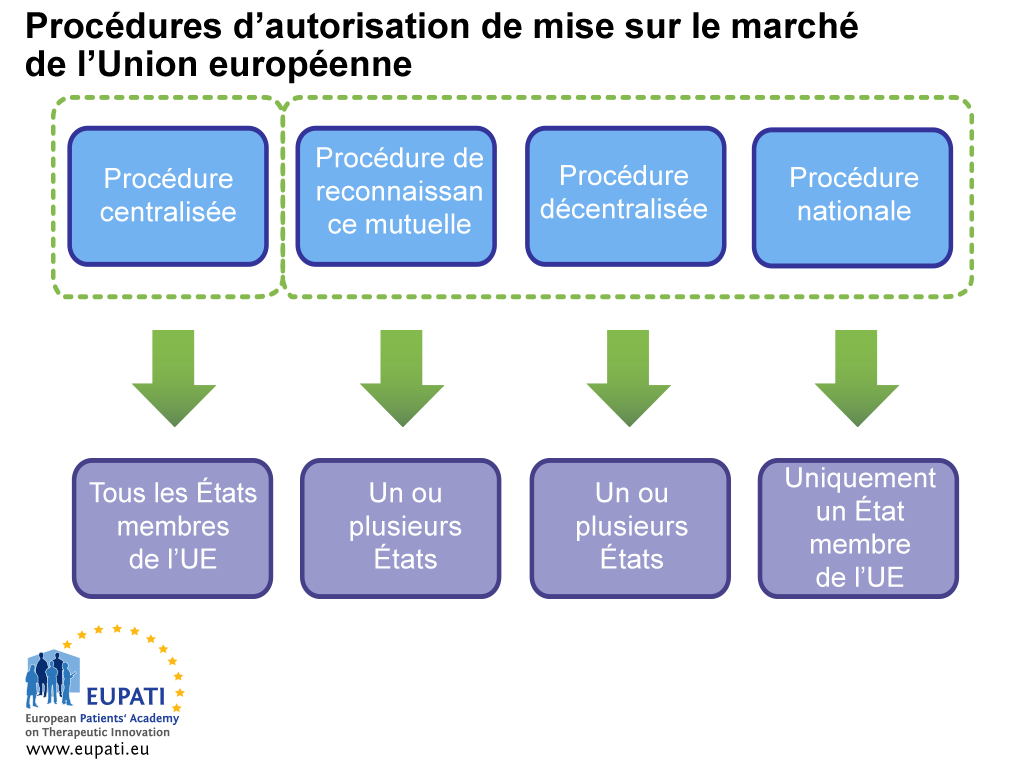
Procédures d’autorisation de mise sur le marché
Une AMM peut s’obtenir de deux manières différentes : par le biais de la procédure centralisée (CP) ou d’une procédure non centralisée. La dernière solution inclut la procédure décentralisée (DCP), la procédure de reconnaissance mutuelle (MRP) et la procédure nationale. Chaque système dispose de ses propres responsabilités et dispositions juridiques pour l’autorité compétente (EMA ou ANC) et les titulaires d’AMM.
Procédure centralisée (CP)
Il s’agit du cheminement de la plupart des nouveaux médicaments. Le secteur soumet une application unique à l’EMA. Le Comité des médicaments à usage humain (CHMP) de l’EMA (composé d’un membre nommé par chaque État membre de l’UE et pays de l’EEE, et de cinq experts scientifiques) émet sa recommandation quant à l’approbation du médicament auprès de la Commission européenne. Si un médicament est ainsi approuvé, une licence de commercialisation est octroyée pour tous les pays de l’UE et de l’EEE. Ce processus d’évaluation scientifique prend un maximum de 210 jours, mais ce décompte peut être interrompu (suspension connue sous le nom de « clock stop »).
Procédure décentralisée (DCP) et procédure de reconnaissance mutuelle (MRP)
Lors de ces procédures, les autorisations de mise sur le marché sont demandées simultanément auprès de plusieurs États membres, avec un pays prenant la tête en tant qu’État membre de référence (RMS). Si tout se passe bien, le médicament est approuvé pour être commercialisé dans le RMS et les autres pays concernés (États membres concernés ou CMS). Ces procédures sont basées sur le principe de reconnaissance de l’évaluation du RMS par les CMS. La plupart des médicaments génériques sont approuvés via la DCP.
Procédure nationale
Les procédures nationales indépendantes sont strictement limitées aux médicaments qui doivent être autorisés et commercialisés dans un seul État membre (MS). Cette procédure est désormais rarement suivie pour les nouveaux produits.
Sélection d’une procédure d’autorisation de mise sur le marché
Une société pharmaceutique est normalement libre de choisir la procédure à utiliser. Il s’agit d’une décision commerciale qui peut comporter diverses raisons en faveur d’une procédure spécifique. La procédure centralisée est obligatoire pour :
- les produits dérivés des processus biotechnologiques ;
- les médicaments basés sur des thérapies avancées (telles que la thérapie génique, la thérapie cellulaire somatique ou les médicaments issus de l’ingénierie tissulaire) ;
- les produits pharmaceutiques orphelins ;
- les médicaments conçus pour traiter le VIH ou le SIDA, le cancer, les troubles neurodégénératifs, les maladies auto-immunes ou autres maladies du système immunitaire, les maladies virales ou le diabète.
Il est également possible d’utiliser la procédure centralisée pour les médicaments qui constituent des innovations techniques, scientifiques ou thérapeutiques considérables, ou dans l’intérêt de la santé publique, comme par exemple la perte de poids.
Si une société veut développer un médicament existant avec une autorisation de mise sur le marché pour une indication différente, elle doit normalement demander une toute nouvelle autorisation de mise sur le marché.
-
- Différents acteurs sont impliqués dans l’autorisation de mise sur le marché d’un médicament selon la procédure choisie par le promoteur ou celle qu’il est obligé de suivre.
Évaluation scientifique des médicaments
Quelle que soit la procédure, chaque médicament doit avoir un « dossier » standard d’évaluation d’autorisation de mise sur le marché. Le Document technique commun (CTD) est un format harmonisé reconnu internationalement qui doit être suivi pour les demandes devant être présentées aux autorités réglementaires. Ce CTD consiste en un ensemble de documents dans lequel le demandeur doit prouver que le produit pharmaceutique offre la qualité requise, et qu’il est sûr et efficace.
Refus d'autorisation de mise sur le marché
Un refus d’AMM pour un produit pharmaceutique sera motivé par :
- un rapport bénéfices/risques considéré comme non favorable ; ou
- une efficacité thérapeutique insuffisamment justifiée par le demandeur ; ou
- une composition qualitative ou quantitative différente de celle déclarée.
Validité de l'autorisation de mise sur le marché et suivi
L’AMM est renouvelée après cinq ans sur la base d’une réévaluation du rapport bénéfices/risques par l’autorité compétente de l’État membre responsable. Une fois renouvelée, l’AMM est valide pour une période illimitée, sauf si l’autorité compétente décide sur la base d’arguments de pharmacovigilance justifiés de procéder à un renouvellement supplémentaire de cinq ans.
Tous les titulaires d’autorisation de mise sur le marché (TAMM) doivent fournir des données de sécurité de suivi et observer d’autres exigences post-commercialisation. Une fois sur le marché, les médicaments doivent en fait continuer d’être surveillés (pharmacovigilance) pour s’assurer que tout aspect susceptible d’en affecter le profil de sécurité est détecté et évalué, et que les mesures nécessaires sont prises.
Pendant qu’un médicament est commercialisé, de nouvelles données ou informations sur son efficacité, sa sécurité ou sa production peuvent devenir disponibles. Dans ce cas, il incombe au TAMM d’envoyer une demande de « variation »d’une autorisation valide de mise sur le marché aux autorités compétentes de tous les États membres qui ont précédemment autorisé le produit en question.
Implication des patients dans l’autorisation de mise sur le marché
L’implication des patients dans l’autorisation de mise sur le marché a traditionnellement été très réduite. Néanmoins, certaines autorités compétentes examinent comment impliquer les patients dans les procédures d’autorisation de mise sur le marché, par le biais par exemple de la consultation des patients ou par une représentation des patients dans la Commission sur les médicaments à usage humain au sein de l’Agence de réglementation des médicaments et des produits de santé (MHRA) au Royaume-Uni. Les patients sont déjà impliqués dans les discussions sur les bénéfices et les risques au niveau de l’EMA en tant que membres du Comité d'évaluation des risques en matière de pharmacovigilance (PRAC), par le biais de leur participation aux procédures de conseil scientifique/assistance au protocole, et lors des réunions de Groupes consultatifs scientifiques (SAG). Il existe également une initiative pilote impliquant les patients dans le CHMP pendant les explications orales précédant la prise de décision.
Les défenseurs des patients ont également la possibilité d’influencer la législation nationale/européenne au sujet de l’autorisation de mise sur le marché.
Références
- The European Economic Area (EEA) unites the EU Member States and the three EEA EFTA States (Iceland, Liechtenstein, and Norway) into the internal market governed by the same basic rules. These rules aim to enable goods, services, capital, and persons to move freely about the EEA in an open and competitive environment, a concept referred to as the four freedoms.
Annexes
A2-5.07-v1.1