

Introduzione
Sono necessari più di 12 anni e in media più di 1 miliardo di euro per condurre tutte le ricerche e lo sviluppo richiesti prima che un nuovo medicinale sia disponibile per l’uso da parte dei pazienti.
Lo sviluppo di farmaci è un’impresa ad alto rischio. La maggioranza (circa il 98%) delle sostanze sviluppate non riesce ad arrivare sul mercato come nuovo medicinale. Ciò accade soprattutto perché osservando i benefici e i rischi (effetti collaterali negativi) riscontrati durante lo sviluppo, non sono comparabili positivamente rispetto ai farmaci già disponibili per i pazienti.
Lo sviluppo di un nuovo farmaco può essere suddiviso in 10 fasi diverse. Il seguente articolo si occupa della Fase 6: prova del meccanismo – studi clinici di Fase I.
-
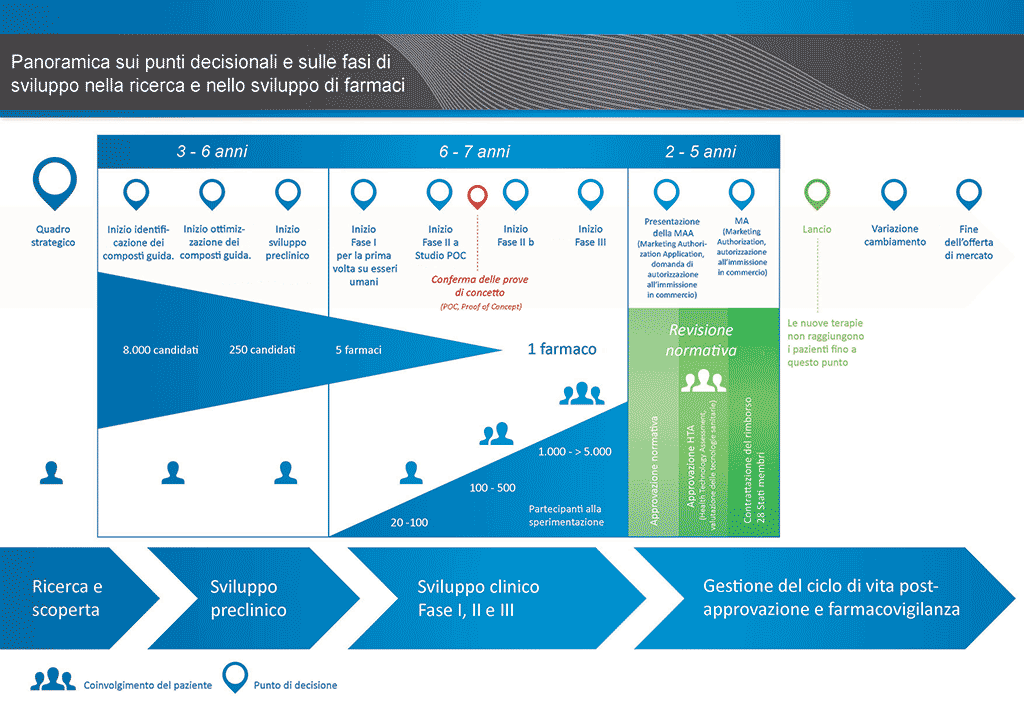
- Occorrono oltre 10 anni di attenta pianificazione e ricerca perché un farmaco passi da molecola a trattamento disponibile sul mercato.
Fase 6: Prova del meccanismo – Studi clinici di Fase I
La decisione di avviare il primo studio clinico è di rilevante entità. Mano a mano che il composto candidato prosegue il processo di sviluppo, aumentano sia il numero, sia il costo, sia la complessità delle attività che il processo comporta.
Prima di avviare uno studio clinico, deve essere presentata una domanda di autorizzazione alla sperimentazione clinica (CTA, Clinical Trial Application). La CTA deve includere i seguenti importanti documenti:
- Un dossier del prodotto medicinale sperimentale (IMPD, Investigational Medicinal Product Dossier), che include ADME e studi che osservano gli effetti (sul target), la sicurezza tossicologica e informazioni sul modo in cui è fabbricato il farmaco.
- Il protocollo di studio, che descrive informazioni dettagliate sull'esecuzione dello studio e sulla valutazione dei risultati.
- Il dossier per lo sperimentatore (IB, Investigator's Brochure): fornisce un riassunto dei dati che permette ai medici che conducono lo studio (gli sperimentatori) di comprendere il modo in cui il farmaco di studio agisce nell'organismo (farmacologia). Esso permette agli sperimentatori di spiegare lo studio ai volontari o ai pazienti e di ottenere il consenso informato (vedere sotto).
La CTA deve essere presentato all'autorità nazionale competente (NCA, National Competent Authority) per l'approvazione. Durante il processo, viene richiesto anche un parere da parte del comitato etico.
La sicurezza è la priorità principale; uno studio negli esseri umani non può iniziare fino a che il comitato di revisione interno dell'azienda, il comitato etico esterno e l'autorità di regolamentazione esterna non hanno dato la loro approvazione.
Studi su volontari (chiamati studi esplorativi, studi di prova del meccanismo o studi di Fase I)
Questo studio permette ai medici e agli scienziati di osservare se il farmaco è sicuro in esseri umani. Osserva anche se il farmaco si comporta negli esseri umani allo stesso modo in cui si comportava negli animali. Esso fornisce informazioni sul modo in cui il farmaco agisce – chiamato "meccanismo d'azione". Tali studi hanno come scopo scoprire anche eventuali effetti secondari del farmaco.
Nella Fase I degli studi clinici sono inclusi circa 20–100 volontari. Questi studi sono di solito condotti in speciali unità di Fase I in cui sono reclutati i volontari. I medici che effettuano questi studi sono chiamati sperimentatori e sono qualificati per condurre studi clinici per determinare il risultato dello studio.
Il primo studio clinico viene di solito condotto in volontari sani di sesso maschile. I dettagli dello studio clinico devono essere descritti nel protocollo di studio e devono includere:
- il background della malattia (il bisogno insoddisfatto),
- le informazioni di tipo non clinico,
- i dettagli dello studio clinico (cosa sarà fatto esattamente e quando) e
- il modo in cui le informazioni saranno utilizzate e analizzate.
Tutte le informazioni provenienti dallo studio vengono inserite in una scheda raccolta dati (CRF, Case Record Form).
Anche in questo caso, esiste un gran numero di linee guida e regolamenti noti come Buona pratica clinica (GCP, Good Clinical Practice), con lo scopo di proteggere la sicurezza dei partecipanti allo studio.
Il protocollo di studio ha anche una sezione sulla "statistica", che contiene i test statistici utilizzati per analizzare i risultati. Tali orientamenti devono essere decisi prima che inizi lo studio; così sarà noto il modo in cui le informazioni saranno ottenute e utilizzate una volta terminato lo studio.
Due elementi molto importanti sono:
- il consenso informato (che assicura che i partecipanti comprendano cosa sarà fatto e acconsentano a partecipare allo studio) e
- la revisione e il parere del comitato etico.
Il Comitato etico è un gruppo indipendente, di solito composto da medici, scienziati, personale infermieristico e non esperti (membri "laici"). Prima che lo studio sia effettuato, riesaminano il protocollo di studio (in particolare il modulo di consenso informato) e si assicurano che sia conforme alle norme etiche del comitato. La sicurezza è la priorità principale e, per garantire quella dei partecipanti a uno studio clinico, sono necessari l'approvazione interna dell'azienda, il parere positivo del comitato etico e l'approvazione dell'autorità nazionale competente (NCA, National Competent Authority). Le norme per gli studi di Fase I sono diventate ancora più severe dopo un caso inusuale, nel 2006, in cui dei volontari hanno sofferto di gravi effetti collaterali utilizzando un farmaco immunomodulante per il trattamento della leucemia linfocitica cronica delle cellule B e dell'artrite reumatoide.
Poiché la sicurezza è una priorità, il primo studio clinico inizia con una dose molto bassa di farmaco:
- Per ciascun volontario viene utilizzata una dose singola di farmaco.
- Una volta dimostrato che con la prima dose non ci sono problemi riguardo alla sicurezza, lo studio può continuare con una dose leggermente più elevata.
- La dose sarà poi ulteriormente aumentata ("dose crescente") fino a che non viene raggiunta la dose massima consentita per lo studio.
Tutto questo viene descritto nel protocollo di studio.
I risultati possono essere poi analizzati ed è possibile determinare tutte le misure riguardanti la sicurezza. Ciò include:
- la farmacocinetica: in che modo l'organismo agisce sul farmaco. Possono essere misurati i livelli del farmaco nel sangue, per determinare Assorbimento, Distribuzione, Metabolismo ed Escrezione (ADME, Absorption, Distribution, Metabolism and Excretion).
- la farmacodinamica: in che modo il farmaco agisce sull'organismo (l'"effetto"). Ad esempio, lo studio potrebbe misurare l'effetto di una medicina su certe cellule ematiche.
Questo tipo di studio è chiamato studio a dose singola crescente (SAD, Single Ascending Dose). Di solito è seguito da uno studio a dose multipla crescente (MAD, Multiple Ascending Dose) che, come il nome suggerisce, comporta dosi multiple per ogni volontario.
A parte gli studi con SAD e MAD, sono necessari anche altri studi di Fase I. Ad esempio:
- per osservare gli effetti del cibo;
- per osservare gli effetti di altri farmaci somministrati contemporaneamente;
- per osservare gli effetti di altre malattie per cui potrebbe essere necessaria una dose diversa di medicinale (per esempio, in pazienti con malattia renale).
Riferimenti bibliografici
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford, UK: Wiley-Blackwell.
Allegati
- Scheda informativa: Studi di prova del meccanismo
Size: 100,054 bytes, Format: .docx
Questa scheda informativa si occupa degli studi clinici di fase I o studi di prova del meccanismo, i primi ad esaminare un candidato composto in esseri umani.
- Presentazione: i principi di base relativi alla scoperta e allo sviluppo di farmaci
Size: 877,906 bytes, Format: .pptx
I principi di base relativi alla scoperta e allo sviluppo di farmaci. Sono necessari più di 12 anni e più di 1 miliardo di euro per condurre tutte le ricerche e lo sviluppo richiesti prima che un nuovo medicinale sia disponibile per l’uso da parte dei pazienti. Questa presentazione spiega in dettaglio il percorso a partire dalla scoperta fino all’immissione sul mercato di un nuovo farmaco e oltre.
A2-1.02.5-v1.1