Last update: 3 augustus 2015


Inleiding
Gemiddeld duurt het ruim 12 jaar en kost het meer dan 1 miljard euro om al het benodigde onderzoeks- en ontwikkelingswerk te doen voordat een nieuw geneesmiddel beschikbaar komt om door patiënten te worden gebruikt.
Geneesmiddelenontwikkeling is een risicovolle onderneming. De meeste stoffen (ongeveer 98%) die worden ontwikkeld, halen de markt niet als nieuw geneesmiddel. Dit komt voornamelijk doordat de voordelen en risico’s (negatieve bijwerkingen) die tijdens de ontwikkeling worden geconstateerd, zich slecht verhouden tot geneesmiddelen die al verkrijgbaar zijn voor patiënten.
De ontwikkeling van een nieuw geneesmiddel kan worden onderverdeeld in 10 verschillende stappen. Het volgende artikel gaat over Stap 6: Proof of mechanism – Klinische onderzoeken in Fase I.
-
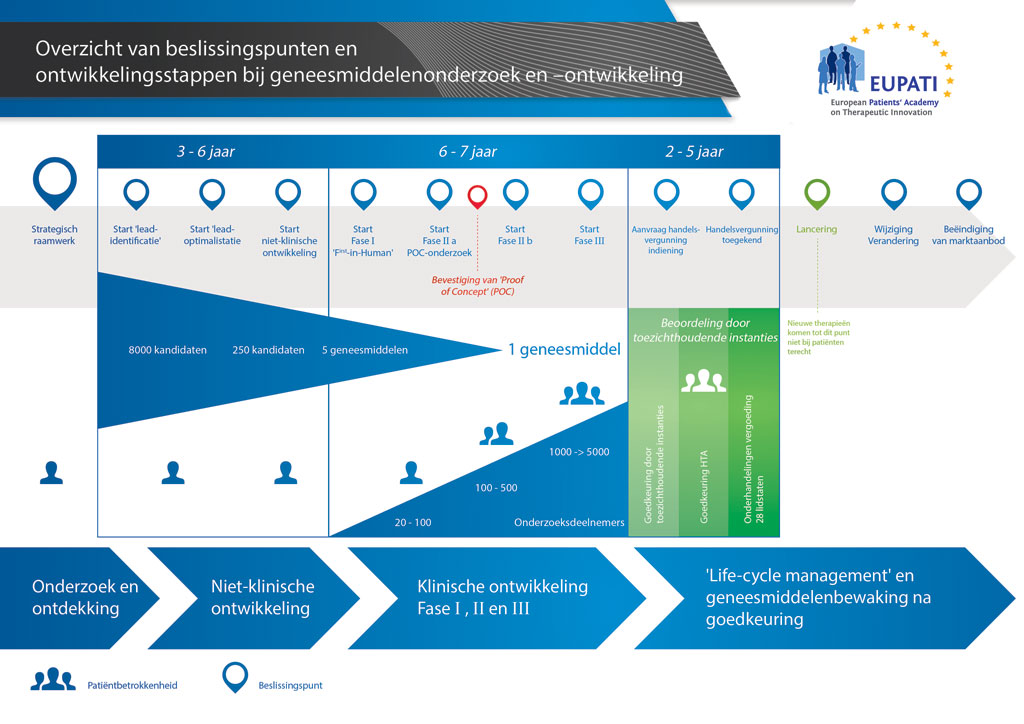
- Er kunnen meer dan 10 jaar aan nauwkeurig plannen en onderzoek nodig zijn om een geneesmiddel te ontwikkelen van een molecuul tot een verkoopbare behandeling.
Stap 6: Proof of mechanism – Klinische onderzoeken in Fase I
De beslissing om het eerste klinische onderzoek te starten is een belangrijke beslissing. Naarmate een kandidaat-stof verder in het ontwikkelingsproces komt, nemen het aantal, de kosten en de complexiteit van de activiteiten die bij het proces horen allemaal toe.
Voordat een klinisch onderzoek kan starten, moet eerst een ‘aanvraag voor een klinische proef’ worden ingediend. De aanvraag moet de volgende belangrijke documenten bevatten:
- Een Investigational Medicinal Product Dossier (IMPD), inclusief ADME en onderzoeken om het effect (op het doelwit) te observeren, de toxicologische veiligheidsinformatie, en informatie over hoe het geneesmiddel is vervaardigd.
- Het onderzoeksprotocol, waarin gedetailleerd staat beschreven hoe het onderzoek wordt uitgevoerd en de resultaten worden geëvalueerd
- De Investigator’s Brochure (IB), met een samenvatting van de informatie zodat de artsen die het onderzoek uitvoeren (de onderzoekers) begrijpen hoe het onderzoeksgeneesmiddel werkt in het lichaam (de farmacologie). Dit stelt de onderzoekers in staat om het onderzoek uit te leggen aan de vrijwilliger of patiënt en geïnformeerde toestemming te krijgen (zie hieronder).
De aanvraag moet voor goedkeuring worden ingediend bij de nationale bevoegde autoriteit. Tijdens de procedure zal ook het advies van de ethische commissie worden ingewonnen.
Veiligheid is de topprioriteit; daarom kan een onderzoek met mensen niet starten voordat de interne beoordelingscommissie van het bedrijf, de externe ethische commissie en de externe toezichthoudende autoriteit hun goedkeuring hebben verleend.
Onderzoeken met vrijwilligers (ook aangeduid als verkennende studies, ‘proof of mechanism’-onderzoeken of fase I-onderzoeken.)
Dit onderzoek stelt artsen en wetenschappers in staat om te zien of het geneesmiddel veilig is bij mensen. Er wordt ook gekeken of het geneesmiddel zich bij mensen op dezelfde manier gedraagt als bij dieren. Deze onderzoeken verschaffen informatie over de manier waarop een geneesmiddel werkt – het zogeheten ‘werkingsmechanisme’. Met deze onderzoeken wordt ook getracht eventuele secundaire effecten van het geneesmiddel te ontdekken.
Er worden ongeveer 20-100 vrijwilligers opgenomen in de eerste fase van de klinische onderzoeken. Deze onderzoeken worden meestal uitgevoerd op speciale fase I-afdelingen waar de vrijwilligers worden gerekruteerd en de onderzoeken worden uitgevoerd. De artsen die deze onderzoeken uitvoeren, worden onderzoekers genoemd en ze zijn gekwalificeerd om klinische onderzoeken uit te voeren om de uitkomst van het onderzoek te bepalen.
Het eerste klinische onderzoek wordt meestal gedaan met gezonde mannelijke vrijwilligers. De gedetailleerde informatie over het klinische onderzoek staat beschreven in het onderzoeksprotocol en moet de volgende onderdelen bevatten:
- de achtergrond van de ziekte (de onvervulde behoefte),
- de niet-klinische informatie,
- gedetailleerde informatie over het klinische onderzoek (wat er precies wordt gedaan en wanneer), en
- hoe de informatie zal worden gebruikt en geanalyseerd.
Alle informatie uit het onderzoek wordt verzameld in een document genaamd ‘case report form’ (CRF).
Ook hier gelden vele richtlijnen en voorschriften, bekend als goede klinische praktijken (Good Clinical Practice, GCP), om de veiligheid van de deelnemers in het onderzoek te beschermen.
Het onderzoeksprotocol bevat ook een rubriek over ‘statistieken’, dat zijn de statistische toetsen die worden gebruikt om de resultaten te analyseren. Deze route moet worden overeengekomen voordat het onderzoek begint, zodat het bekend is hoe de informatie wordt verkregen en gebruikt als het onderzoek is afgelopen.
Twee zeer belangrijke elementen zijn:
- Geïnformeerde toestemming (waarborgen dat de deelnemers begrijpen wat er zal worden gedaan en akkoord gaan met deelname aan het onderzoek), en
- Beoordeling en advies van de ethische commissie.
De ethische commissie is een onafhankelijke groep, meestal bestaande uit artsen, wetenschappers, verpleegkundigen en niet-deskundigen (leken). Ze beoordelen het onderzoeksprotocol (met name het toestemmingsformulier) en letten erop dat alles voldoet aan de ethische voorschriften van de commissie voordat het onderzoek wordt uitgevoerd. Veiligheid heeft de hoogste prioriteit; om de veiligheid van de deelnemers in een klinisch onderzoek te waarborgen, is goedkeuring vereist van het bedrijf zelf, van een externe ethische commissie en van de nationale bevoegde autoriteit. De regels voor Fase I-onderzoeken werden nog strenger na een zeldzaam geval in 2006 waarbij vrijwilligers ernstige bijwerkingen kregen na gebruik van een immunomodulerend geneesmiddel voor de behandeling van B-cel chronische lymfatische leukemie en reumatoïde artritis.
Omdat veiligheid een prioriteit is, begint het eerste klinische onderzoek met een zeer lage dosis van het geneesmiddel:
- Voor elke vrijwilliger wordt een enkelvoudige dosis van het geneesmiddel gebruikt.
- Als eenmaal is aangetoond dat er geen veiligheidskwesties zijn met deze eerste dosis, kan het onderzoek verdergaan met een iets hogere dosis.
- De dosis zal vervolgens worden verhoogd (‘oplopende doses’) tot de maximale dosis die voor dit onderzoek is toegestaan, is bereikt.
Dit staat beschreven in het onderzoeksprotocol.
Daarna kunnen de onderzoeksresultaten worden geanalyseerd en alle veiligheidsmetingen beoordeeld. Dat zijn:
- farmacokinetiek – wat het lichaam doet met het geneesmiddel. De bloedspiegels van het geneesmiddel kunnen worden gemeten, om absorptie, distributie, metabolisme en excretie (ADME) van een geneesmiddel te bepalen
- farmacodynamiek – wat het geneesmiddel doet met het lichaam (het ‘effect’). Bijvoorbeeld, het onderzoek kan het effect van het geneesmiddel op bepaalde bloedcellen meten.
Dit type onderzoek wordt een onderzoek met oplopende enkelvoudige doses genoemd. Hierna volgt meestal een onderzoek met oplopende meervoudige doses dat, zoals de naam al suggereert, meerdere doses per vrijwilliger behelst.
Naast de onderzoeken met oplopende enkelvoudige doses en oplopende meervoudige doses zijn er ook andere Fase I-onderzoeken nodig. Bijvoorbeeld:
- om te kijken naar het effect van voeding
- om te kijken naar het effect van andere geneesmiddelen die tegelijkertijd worden gegeven
- om te kijken naar het effect van andere ziekten die ertoe kunnen leiden dat een afwijkende dosis van het geneesmiddel nodig is (bijvoorbeeld bij patiënten met een nierziekte).
Referenties
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford, UK: Wiley-Blackwell.
Bijlagen
- Factsheet: ‘Proof of mechanism’-onderzoeken
Size: 98,674 bytes, Format: .docx
Dit factsheet gaat over klinische of ‘proof of mechanism’-onderzoeken in fase I, de eerste onderzoeken die een kandidaat-stof bij mensen testen.
- Presentatie: De basisprincipes van geneesmiddelontdekking en -ontwikkeling
Size: 950,426 bytes, Format: .pptx
De basisprincipes van geneesmiddelontdekking en -ontwikkeling. Gemiddeld duurt het ruim 12 jaar en kost het meer dan 1 miljard euro om al het benodigde onderzoeks- en ontwikkelingswerk te doen voordat een nieuw geneesmiddel beschikbaar komt om door patiënten te worden gebruikt. Deze presentatie gaat nader in op het proces van ontdekking tot marktintroductie van een nieuw geneesmiddel en daarna.
A2-1.02.5-v1.1