

Einleitung
Es dauert über 12 Jahre und kostet durchschnittlich mehr als eine Milliarde Euro, all die Forschungs- und Entwicklungsarbeiten durchzuführen, die erforderlich sind, bis ein neues Arzneimittel für die Behandlung von Patienten zur Verfügung steht.
Arzneimittelentwicklung ist ein risikoreiches Geschäft. Der größte Teil (ca. 98 %) neu entwickelter Wirkstoffe schafft es nicht, als neues Arzneimittel auf den Markt zu gelangen. Das liegt daran, dass das Verhältnis zwischen dem Nutzen und den im Verlauf der Entwicklung festgestellten Risiken (schädliche Nebenwirkungen) dem Vergleich mit anderen, bereits für die Behandlung von Patienten verfügbaren Arzneimitteln meist nicht standhält.
Die Entwicklung eines neuen Arzneimittels kann in zehn unterschiedliche Schritte unterteilt werden. Der folgende Artikel behandelt Schritt 6: Nachweis des Wirkmechanismus – Klinische Phase-I-Studien.
-
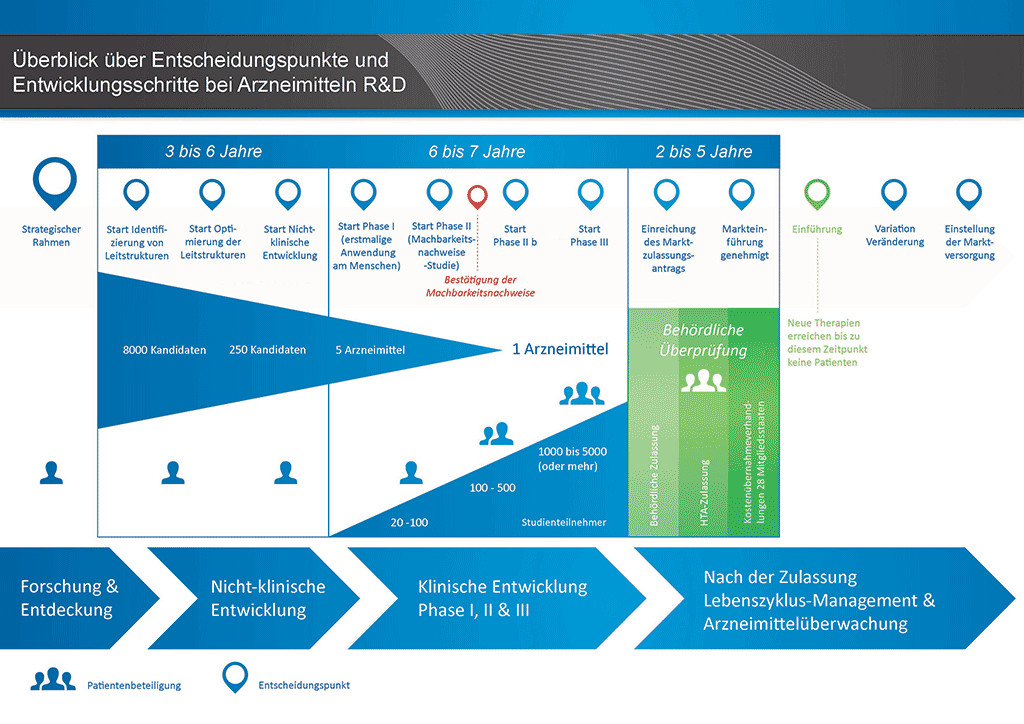
- Es benötigt mehr als 10 Jahre sorgfältiger Planung und Forschung, bis ein Arzneimittel sich vom Molekül zur marktfähigen Behandlung entwickelt hat.
Schritt 6: Nachweis des Wirkmechanismus – Klinische Phase-I-Studien
Die Entscheidung, mit der ersten klinischen Studie zu beginnen, ist eine wesentliche. Wird der Entwicklungsprozess für einen Wirkstoffkandidaten fortgesetzt, erhöhen sich die Anzahl, die Kosten sowie die Komplexität sämtlicher Aktivitäten des Prozesses.
Vor der Aufnahme einer klinischen Studie muss ein Antrag zur klinischen Prüfung (Clinical Trial Application, CTA) eingereicht werden. Dieser Antrag muss die folgenden wichtigen Dokumente enthalten:
- Ein Dossier über die pharmazeutische Qualität des Prüfpräparats (Investigational Medicinal Product Dossier, IMPD); dieses umfasst Informationen zur Pharmakokinetik („ADME“), zu Studien zur Wirkung (auf das „Target“), zur toxikologischen Sicherheit sowie zu den Produktionsverfahren.
- Das Studienprotokoll mit Detailangaben zur Durchführung der Studie und Auswertung der Ergebnisse
- Die Prüferinformation (Investigator's Brochure, IB) mit einer Zusammenfassung der Informationen, die es den leitenden Ärzten der Studie (Prüfern) erlaubt zu verstehen, wie das Prüfpräparat im Körper wirkt (Pharmakologie). Dies erlaubt es den Prüfern, den freiwilligen Probanden oder Patienten die Studie zu erklären, damit diese rechtswirksam in die Teilnahme an der Studie einwilligen können (siehe unten).
Der CTA muss der zuständigen nationalen Behörde zur Genehmigung vorgelegt werden. Während dieses Prozesses wird auch eine Stellungnahme der zuständigen Ethik-Kommission eingeholt.
Sicherheit hat oberste Priorität. Daher kann eine Studie am Menschen erst dann beginnen, wenn der unternehmenseigene Prüfungsausschuss (Internal Company Review Committee), die externe Ethik-Kommission und die externe Aufsichtsbehörde ihre Zustimmung gegeben haben.
Probandenstudien (auch explorative Studien, „Proof-of-Mechanism“-Studien oder Phase-I-Studien genannt)
Diese Studie ermöglicht es Ärzten und Wissenschaftlern, festzustellen, ob das Arzneimittel für Menschen sicher ist. Sie betrachtet auch die Frage, ob sich das Arzneimittel beim Menschen genauso verhält, wie es beim Tier beobachtet wurde. Diese Studien liefern Informationen zum Wirkmechanismus, d. h. der Funktionsweise des Arzneimittels. Sie zielen auch darauf ab, etwaige Nebenwirkungen des Arzneimittels zu finden.
An einer klinischen Phase-I-Studie sind etwa 20 bis 100 freiwillige Probanden beteiligt. Diese Studien werden normalerweise an speziellen Phase-I-Einrichtungen durchgeführt, wo Freiwillige angeworben und die Studien durchgeführt werden. Die Ärzte, die diese Studien durchführen, werden als „Prüfer“ bezeichnet und sind qualifiziert, klinische Studien durchzuführen und auszuwerten.
Die erste klinische Studie wird normalerweise an gesunden männlichen freiwilligen Probanden durchgeführt. Das Studienprotokoll muss die Details der klinischen Studie beschreiben. Hierzu gehören:
- Hintergrundinformationen zur Krankheit (der unerfüllte Bedarf)
- Die in den nicht-klinischen Untersuchungen gewonnenen Informationen
- Einzelheiten der klinischen Studie (was genau geschehen wird und wann)
- Nutzung und Auswertung der gewonnenen Informationen
Sämtliche im Rahmen der Studie erhobenen und gewonnenen Informationen werden in einem als Prüfbogen (Case Report Form, CRF) bezeichneten Dokument erfasst.
Auch hier gibt es eine Vielzahl von Richtlinien und Bestimmungen – gemeinsam als „Gute klinische Praxis“ (Good Clinical Practice, GCP) bezeichnet –, die die Sicherheit der Teilnehmer an der Studie gewährleisten.
Das Studienprotokoll besitzt außerdem einen Abschnitt „Statistik“, der die statistischen Verfahren vorgibt, mit denen die Ergebnisse analysiert werden. Über diese Vorgaben muss entschieden werden, bevor die Studie beginnt, damit bekannt ist, wie die Informationen am Studienende bestimmt und verwendet werden.
Zwei sehr wichtige Elemente sind:
- Einwilligung nach Aufklärung (die sicherstellt, dass die Teilnehmer sich über die bevorstehenden Geschehnisse im Klaren sind und ihr Einverständnis geben, an der Studie teilzunehmen)
- Bericht und Stellungnahme der Ethik-Kommission
Die Ethik-Kommission ist ein unabhängiges Gremium, das sich aus im Gesundheitswesen und in nichtmedizinischen Bereichen tätigen Personen zusammensetzt. Diese überprüfen vor Aufnahme der Studie das Studienprotokoll (insbesondere die Einverständniserklärung) und stellen sicher, dass es mit den ethischen Bestimmungen des Ausschusses übereinstimmt. Sicherheit hat oberste Priorität; um die Sicherheit der Teilnehmer einer klinischen Studie sicherzustellen, bedarf es der Zustimmung des unternehmenseigenen Prüfungsausschusses, einer befürwortenden Stellungnahme der externen Ethik-Kommission und der Freigabe durch die zuständige Aufsichtsbehörde. Die Richtlinien für Phase-I-Studien wurden weiter verschärft, nachdem 2006 nach der Einnahme eines immunmodulatorischen Wirkstoffs für die Behandlung von chronisch-lymphatischer B-Zell-Leukämie und rheumatoider Arthritis bei freiwilligen Probanden schwerwiegende Nebenwirkungen auftraten. (Es sei darauf hingewiesen, dass es sich hierbei um eine absolute Ausnahme handelte.)
Da Sicherheit Priorität hat, beginnt die erste klinische Studie mit einer sehr niedrigen Dosis des Arzneimittels:
- Jeder Proband erhält eine Einzeldosis des Arzneimittels.
- Sobald feststeht, dass es keine Sicherheitsbedenken mit dieser ersten Dosis gibt, kann die Studie mit einer etwas höheren Dosis fortgesetzt werden.
- Die Dosis wird dann weiter erhöht („Dosiseskalation“), bis die maximale für die Studie zugelassene Dosis erreicht worden ist.
Diese Vorgehensweise ist im Studienprotokoll beschrieben.
Die Studienergebnisse können dann analysiert und alle Erkenntnisse zur Sicherheit beurteilt werden. Dies umfasst:
- Pharmakokinetik – Was der Körper mit dem Arzneimittel macht. Der Arzneimittelspiegel im Blut kann gemessen und auf diese Weise die Pharmakokinetik (Resorption, Verteilung, Verstoffwechselung und Ausscheidung) bestimmt werden.
- Pharmakodynamik – Was das Arzneimittel mit dem Körper macht (die „Wirkung“). Die Studie könnte z. B. die Wirkung eines Arzneimittels auf bestimmte Blutzellen messen.
Studien dieser Art werden als Einzeldosis-Eskalationsstudie (Single Ascending Dose, SAD-Studie) bezeichnet. Auf diese folgt üblicherweise eine Mehrfachdosis-Eskalationsstudie ((Multiple Ascending Dose, MAD-Studie), die, wie der Name besagt, mehrere Dosen je Proband vorsieht.
Neben SAD- und MAD-Studien werden auch andere Phase-I-Studien benötigt. Zum Beispiel:
- Betrachtung der Auswirkung von Nahrung
- Betrachtung der Auswirkung anderer, gleichzeitig eingenommener Arzneimittel
- Betrachtung der Auswirkung anderer Krankheiten, die möglicherweise eine Anpassung der Dosis des Arzneimittels erforderlich machen (z. B. bei Patienten mit Nierenerkrankungen)
Quellenangaben
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford, UK: Wiley-Blackwell.
Anlagen
- Datenblatt: Studien zum Nachweis des Wirkmechanismus
Size: 100,313 bytes, Format: .docx
Dieses Datenblatt behandelt klinische Phase-I-Studien (Studien zum Nachweis des Wirkmechanismus). Hierbei handelt es sich um die ersten Studien, bei denen ein Wirkstoffkandidat am Menschen getestet wird.
- Präsentation: Die grundlegenden Prinzipien der Arzneimittelentdeckung und -entwicklung
Size: 945,895 bytes, Format: .pptx
Die grundlegenden Prinzipien der Arzneimittelentdeckung und -entwicklung. Es dauert über 12 Jahre und kostet mehr als eine Milliarde Euro, all die Forschungs- und Entwicklungsarbeiten durchzuführen, die erforderlich sind, bis ein neues Arzneimittel für die Behandlung von Patienten zur Verfügung steht. Diese Präsentation stellt die Details des Prozesses von der Entdeckung bis zur Markteinführung eines neuen Arzneimittels und darüber hinaus vor.
A2-1.02.5-v1.1