Last update: 3 августа 2015


Введение
В среднем на все исследования и разработки, необходимые для того, чтобы новый лекарственный препарат был доступен для пациентов, уходит более 12 лет и более 1 миллиарда евро.
Разработка лекарственных препаратов — это рисковый бизнес. Большинство разрабатываемых соединений (около 98 %) так и не выходят на рынок. Так происходит потому, что при оценке преимуществ и рисков (негативных побочных эффектов), обнаруживаемых в ходе разработки, сложно сравнивать их с уже имеющимися на рынке препаратами.
Процесс разработки нового лекарственного препарата можно представить в 10 шагах. Следующая статья описывает 6-й шаг. Доказательство механизма действия — клинические исследования 1 фазы
-
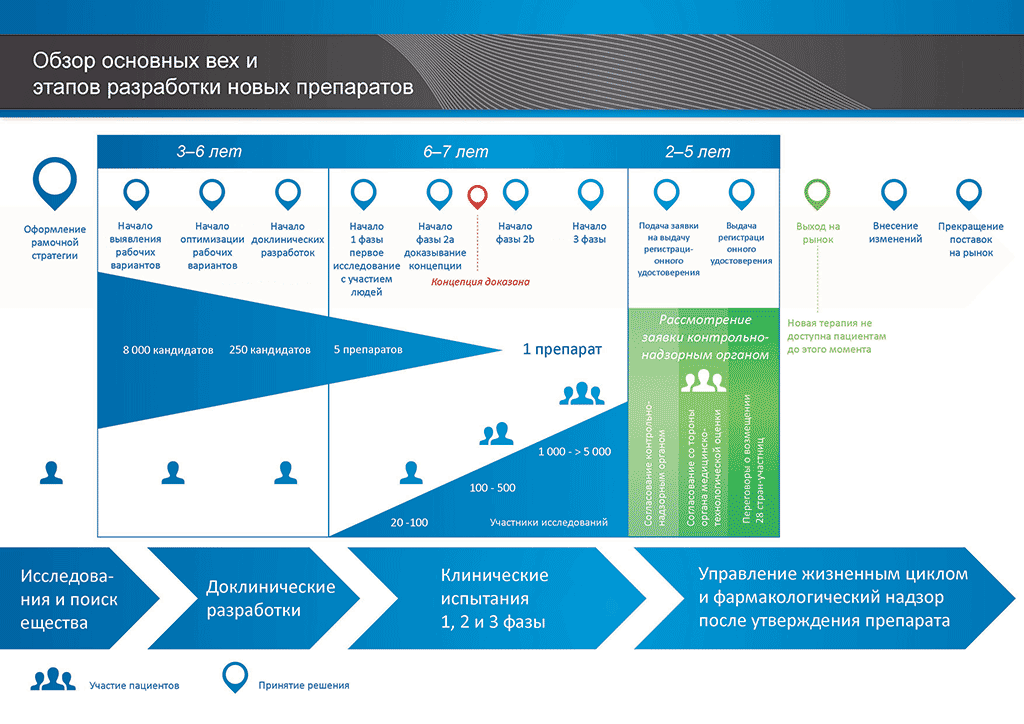
- С момента создания молекулы до момента начала продажи медицинского препарата проходит больше 10 лет, необходимых для тщательнейшего планирования и исследований.
-
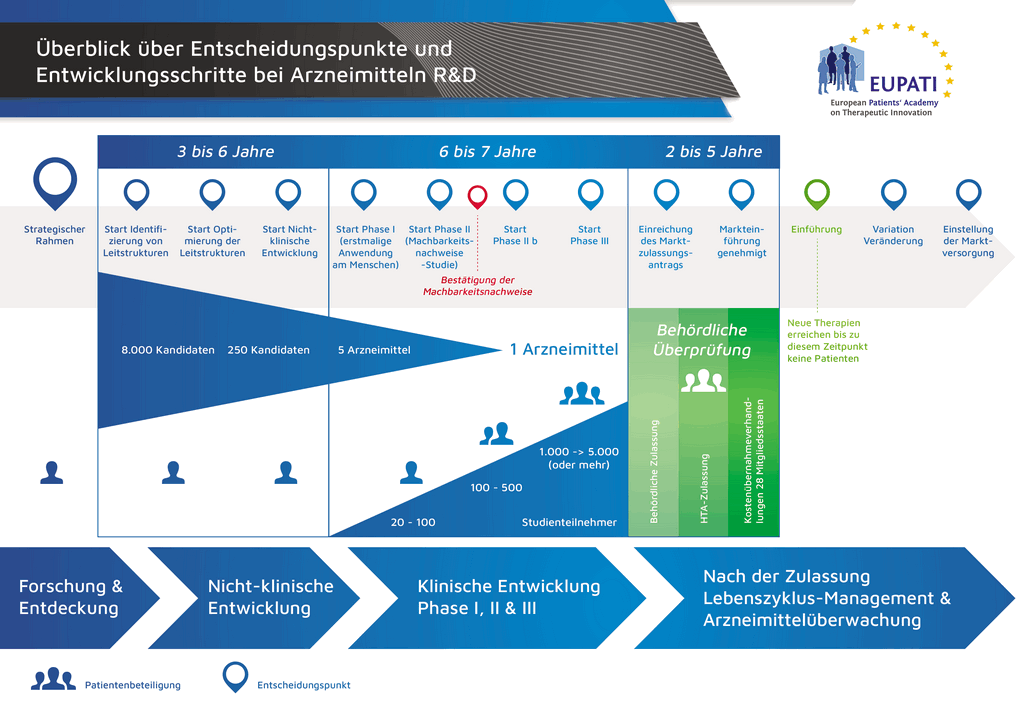
- Es benötigt mehr als 10 Jahre sorgfältiger Planung und Forschung, bis ein Arzneimittel sich vom Molekül zur marktfähigen Behandlung entwickelt hat.
Шаг 6: Доказательство механизма действия — клинические исследования 1 фазы
Важной вехой является принятие решения о проведении первого клинического исследования. По мере того, как разворачиваются новые этапы разработки исследуемого вещества, увеличивается количество, стоимость и сложность проводимых в процессе разработки мероприятий.
До начала клинического исследования необходимо подать заявку на проведение клинического исследования (CTA). К ней прилагаются следующие важные документы:
- досье исследуемого лекарственного препарата (IMPD), включая данные о всасывании, распределении, метаболизме и выведении препарата (ADME) и исследованиях эффекта (на мишень), данные о токсикологической безопасности, а также информацию о производстве препарата;
- протокол исследования, в котором описываются подробности проведения исследования и оценки результатов;
- брошюра исследователя (IB), в которой обобщается информация, позволяющая проводящим исследование врачам (исследователям) понять, как исследуемый лекарственный препарат действует в организме (фармакология). Это позволяет исследователям объяснить суть исследования волонтерам и пациентам и получить их информированное согласие (см. ниже).
Заявка на проведение клинического исследования подается в национальный компетентный орган для согласования. В рамках этого процесса также запрашивается мнение комитета по этике.
Безопасность является задачей номер один, в связи с чем исследование с участием людей не может быть начато, пока внутренний наблюдательный комитет компании, независимый комитет по этике и независимый контрольно-надзорный орган не согласуют его проведение.
Исследование с участием волонтеров (также называется «поисковым исследованием», «исследованием с целью доказательства механизма действия» и «исследованием 1 фазы»).
Проведение этого исследования позволяет врачам и ученым понять, безопасно ли применение препарата людьми. Также оно помогает понять, действует ли препарат на людей так же, как и на животных. Эти исследования позволяют получить данные о том, как действует препарат — о «механизме действия». Кроме того, эти исследования направлены на получение данных о вторичных эффектах препарата.
В клинических исследованиях 1 фазы принимают участие 20–100 волонтеров. Обычно эти исследования проводятся в специальных отделениях, где набирают волонтеров и проводят исследования. Врачи, проводящие эти исследования, называются «исследователями», и они обладают нужной квалификацией для проведения клинических испытаний с целью определения результатов исследования.
Первое клиническое исследование обычно проводится с участием здоровых мужчин-волонтеров. Подробности клинического исследования должны быть описаны в протоколе исследования, при этом они включают:
- сведения об истории заболевания (неудовлетворенной потребности);
- доклинические данные;
- подробное описание клинического исследования (что конкретно и когда будет происходить); а также
- способы использования и анализа данных.
Все сведения, поступающие в ходе исследования, протоколируются в документе «индивидуальная регистрационная карта» (CRF).
Существует также развернутый свод норм и правил, обеспечивающих безопасность участников исследования, известный как «надлежащая клиническая практика» (GCP).
В протоколе исследования также включен раздел, посвященный статистике, в котором описываются статистические тесты, используемые для анализа результатов. Эти нюансы необходимо определить до начала исследования, чтобы знать, как будет собираться и использоваться информация после окончания исследования.
Следующие два момента очень важны:
- информированное согласие (обеспечивает понимание участниками того, что будет происходить, и их согласие на участие в исследовании); и
- оценка и заключение комитета по этике.
Комитет по этике представляет из себя независимую группу врачей, ученых, медперсонала и лиц, не являющихся специалистами (представителей общественности). Они изучают протокол исследования (в особенности форму информированного согласия), чтобы до начала исследования удостовериться в его соответствии нормам комитета по этике. Безопасность — задача номер один, поэтому в целях безопасности участников клинического исследования необходимо согласование со стороны самой компании, независимого комитета по этике и национального компетентного органа. Правила проведения исследований 1 фазы еще более ужесточились после того, как в 2006 г. произошел редкий случай: у волонтеров, применявших иммуномодулирующее средство для лечения хронического В-клеточного лимфолейкоза и ревматоидного артрита, наблюдались серьезные побочные эффекты.
Так как безопасность является задачей номер один, в первом клиническом исследовании используется очень низкая дозировка препарата.
- Каждый волонтер получает одну дозу препарата.
- Если после применения первой дозы не наблюдается небезопасных явлений, то исследование можно продолжать с повышением дозы.
- Затем доза постепенно повышается до максимально допустимого предела.
Это устанавливается в протоколе исследования.
Затем результаты исследования анализируются и производится оценка всех показателей безопасности препарата. Это включает:
- фармакокинетику — как организм влияет на препарат. Измеряется уровень содержания препарата в крови с целью определения всасывания, распределения, метаболизма и выведения (ADME).
- фармакодинамику — как препарат влияет на организм («эффект»). Например, в исследовании может измеряться эффект препарата на отдельные клетки крови.
Такой тип исследований известен как исследование с однократной нарастающей дозой. Затем обычно проводится исследование с многократными нарастающими дозами, т.е. когда волонтер получает несколько доз препарата.
Помимо исследований с однократной и многократной нарастающей дозами, необходимо проведение прочих исследований 1 фазы. например:
- для изучения влияния пищи на препарат;
- для изучения влияния прочих препаратов, принимаемых одновременно с исследуемым препаратом;
- для изучения влияния прочих заболеваний, когда может потребоваться изменение дозы препарата (например, для пациентов с почечной недостаточностью).
Справочная литература
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford, UK: Wiley-Blackwell.
Приложения
- Информационный бюллетень Исследования, направленные на доказательство механизма действия
Size: 100,641 bytes, Format: .docx
В этом информационном бюллетене описываются клинические исследования 1 фазы, или «исследования с целью доказательства механизма действия», — первые исследования, в ходе которых исследуемое вещество испытывается на людях.
A2-1.02.5-v1.1