Last update: 3 August 2015


Einleitung
Es dauert über 12 Jahre und kostet durchschnittlich mehr als eine Milliarde Euro, all die Forschungs- und Entwicklungsarbeiten durchzuführen, die erforderlich sind, bis ein neues Arzneimittel für die Behandlung von Patienten zur Verfügung steht.
Arzneimittelentwicklung ist ein risikoreiches Geschäft. Der größte Teil (ca. 98 %) neu entwickelter Wirkstoffe schafft es nicht, als neues Arzneimittel auf den Markt zu gelangen. Das liegt daran, dass das Verhältnis zwischen dem Nutzen und den im Verlauf der Entwicklung festgestellten Risiken (schädliche Nebenwirkungen) dem Vergleich mit anderen, bereits für die Behandlung von Patienten verfügbaren Arzneimitteln meist nicht standhält.
Die Entwicklung eines neuen Arzneimittels kann in zehn unterschiedliche Schritte unterteilt werden. Der folgende Artikel behandelt Schritt 5: Nicht-klinische Sicherheitsprüfung.
-
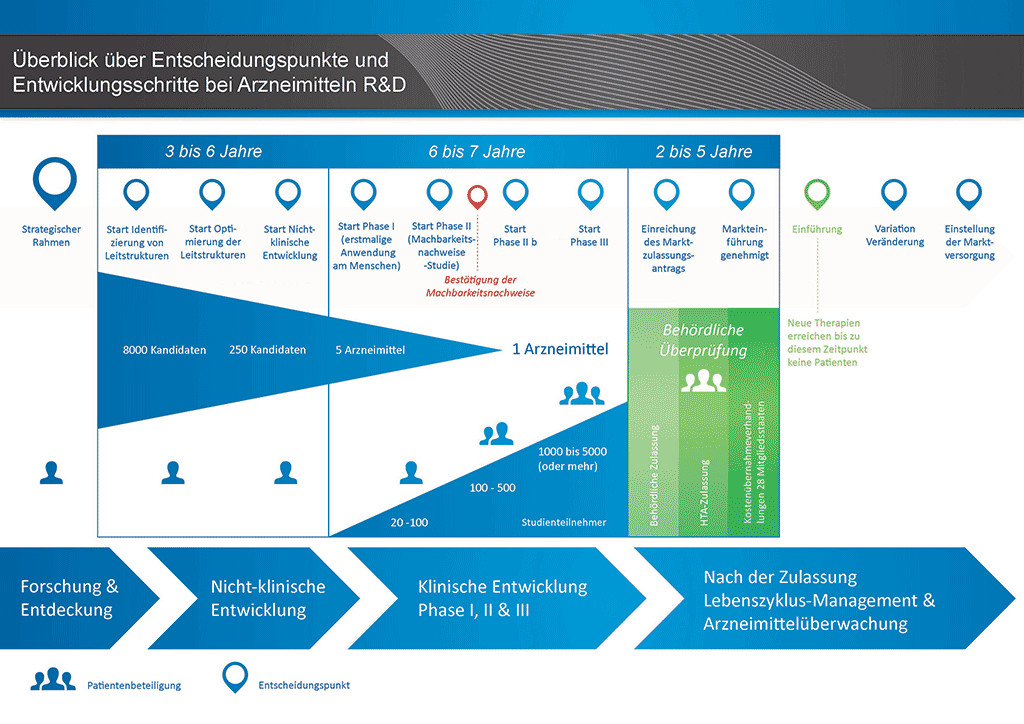
- Es benötigt mehr als 10 Jahre sorgfältiger Planung und Forschung, bis ein Arzneimittel sich vom Molekül zur marktfähigen Behandlung entwickelt hat.
Schritt 5: Nicht-klinische Sicherheitsprüfung
Ist es sicher, mit der klinischen Prüfung fortzufahren? Die nächste Phase im Arzneimittelentwicklungsprozess umfasst Versuche im Tiermodell zur Sicherheit. Diese unterliegen den spezifischen Regeln und Bestimmungen der Guten Laborpraxis (Good Laboratory Practice, GLP). Kein Wirkstoffkandidat kann am Menschen (in „klinischen Studien“) getestet werden, ohne dass zuvor in Tierstudien zur Unbedenklichkeit sein Sicherheitsprofil aufgestellt wurde. Die Arzneimittelentwicklung unterliegt strengen Kontrollen und Reglementierungen. Das Gesetz legt mittels Regeln und Bestimmungen, was zu tun ist und wie es getan wird.
Bevor die nicht-klinischen Untersuchungen durchgeführt werden können, müssen größere Mengen des Wirkstoffkandidaten hergestellt werden, damit sämtliche erforderlichen Tests durchgeführt werden können. Dieser Herstellungsprozess unterliegt ebenfalls strengen Richtlinien und Bestimmungen, die man „Gute Herstellungspraxis“ (Good Manufacturing Practice, GMP) nennt.
Die Bestimmungen legen fest, welche Studien erforderlich sind und an welchen Arten von Tieren diese durchgeführt werden müssen, um angemessene Informationen zu erhalten. Im Rahmen dieser Studien werden die folgenden Auswirkungen des Wirkstoffs ermittelt:
- Gesamtauswirkungen auf das Tier
- Auswirkungen auf sämtliche Gewebe und Organe des Tieres (Untersuchungen zur systemischen Toxizität)
- Untersuchungen auf die Fähigkeit der Tiere, sich fortzupflanzen und sich normal zu entwickeln (Untersuchungen zur Reproduktionstoxizität)
- Auswirkungen auf Haut oder Augen (Untersuchungen zur lokalen Toxizität)
- Auftreten von Allergien (Untersuchungen zur Überempfindlichkeit)
- Auswirkungen auf Chromosomen und Gene (Untersuchungen zur Genotoxizität)
- Jegliche zur Entstehung von Krebs beitragende Effekte (Untersuchungen zur Karzinogenität)
Diese Untersuchungen sind in der nachstehenden Abbildung zu sehen.
Arten von Toxizitätsuntersuchungen
- Untersuchungen zur systemischen Toxizität
- Einzeldosis-Studien
- Mehrfachdosis-Studien
- Untersuchungen zur Reproduktionstoxizität
- Untersuchungen zur männlichen Fertilität
- Untersuchungen zur weiblichen Fertilität und zur Entwicklung der Leibesfrucht
- Untersuchungen zur lokalen Toxizität
- Untersuchungen zur Überempfindlichkeit
- Untersuchungen zur Genotoxizität
- Untersuchungen zur Karzinogenität
Diese Untersuchungen bestimmen nicht nur das Sicherheitsprofil bei Tieren, sondern liefern zusätzlich wichtige Information zu folgenden Aspekten:
- Die Aufnahme des Wirkstoffs im Körper (engl. Absorption)
- Die Verteilung des Wirkstoffs im ganzen Körper (engl. Distribution)
- Der Abbau des Wirkstoffs durch den Körper (engl. Metabolism)
- Die Ausscheidung des Wirkstoffs durch den Körper (engl. Excretion).
Nach den englischen Begriffen werden diese Aspekte zusammenfassend auch mit „ADME“ abgekürzt, im Deutschen ist der Begriff „Pharmakokinetik“ gebräuchlich.
Auf Grundlage all dieser Informationen wird entschieden, ob und ggf. in welcher Dosierung der Wirkstoffkandidat erstmalig am Menschen (klinische Phase-I-Studie) getestet werden kann.
Um zur klinischen Prüfung am Menschen übergehen zu können, muss der Wirkstoffkandidat in allen obligatorischen nicht-klinischen Toxizitätsuntersuchungen ein akzeptables Sicherheitsprofil zeigen. Allerdings werden zu diesem Zeitpunkt noch nicht alle nicht-klinischen Untersuchungen zur Sicherheit abgeschlossen sein. Langzeitstudien zur Karzinogenität beispielsweise nehmen im Durchschnitt zwei Jahre in Anspruch und werden nach Aufnahme der klinischen Studien parallel mit diesen fortgesetzt.
Quellenangaben
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford, UK: Wiley-Blackwell.
Anlagen
- Datenblatt: Arzneimittelentdeckung
Size: 1,226,937 bytes, Format: .docx
Arzneimittelentdeckung. Dieses Datenblatt behandelt die Schritte des Arzneimittelentdeckungs- und -entwicklungsprozesses, die vor dem Test eines Wirkstoffs am Menschen stattfinden – von der Erkenntnisgewinnung (Sammeln von Informationen zu einer Erkrankung) bis hin zu den nichtklinischen Sicherheitstests im Tiermodell.
- Präsentation: Die grundlegenden Prinzipien der Arzneimittelentdeckung und -entwicklung
Size: 945,895 bytes, Format: .pptx
Die grundlegenden Prinzipien der Arzneimittelentdeckung und -entwicklung. Es dauert über 12 Jahre und kostet mehr als eine Milliarde Euro, all die Forschungs- und Entwicklungsarbeiten durchzuführen, die erforderlich sind, bis ein neues Arzneimittel für die Behandlung von Patienten zur Verfügung steht. Diese Präsentation stellt die Details des Prozesses von der Entdeckung bis zur Markteinführung eines neuen Arzneimittels und darüber hinaus vor.
A2-1.02.4-v1.1