Last update: 3 august 2015


Introduktion
Det tager over 12 år og koster i gennemsnit over 1 milliarder kroner at gennemføre al den nødvendige forskning og udvikling, før et nyt lægemiddel er klar til brug for patienter.
Der er høj risiko forbundet med at udvikle lægemidler. De fleste stoffer (omkring 98 %), der udvikles, kommer aldrig på markedet som nye lægemidler. Det skyldes primært, at når man ser på fordelene og risiciene (de negative bivirkninger), i udviklingsfasen, kan de ikke matche de lægemidler, som patienterne allerede har adgang til.
Udviklingen af et nyt lægemiddel kan opdeles i 10 forskellige trin. Følgende artikel dækker trin 5: Prækliniske sikkerhedstest.
-
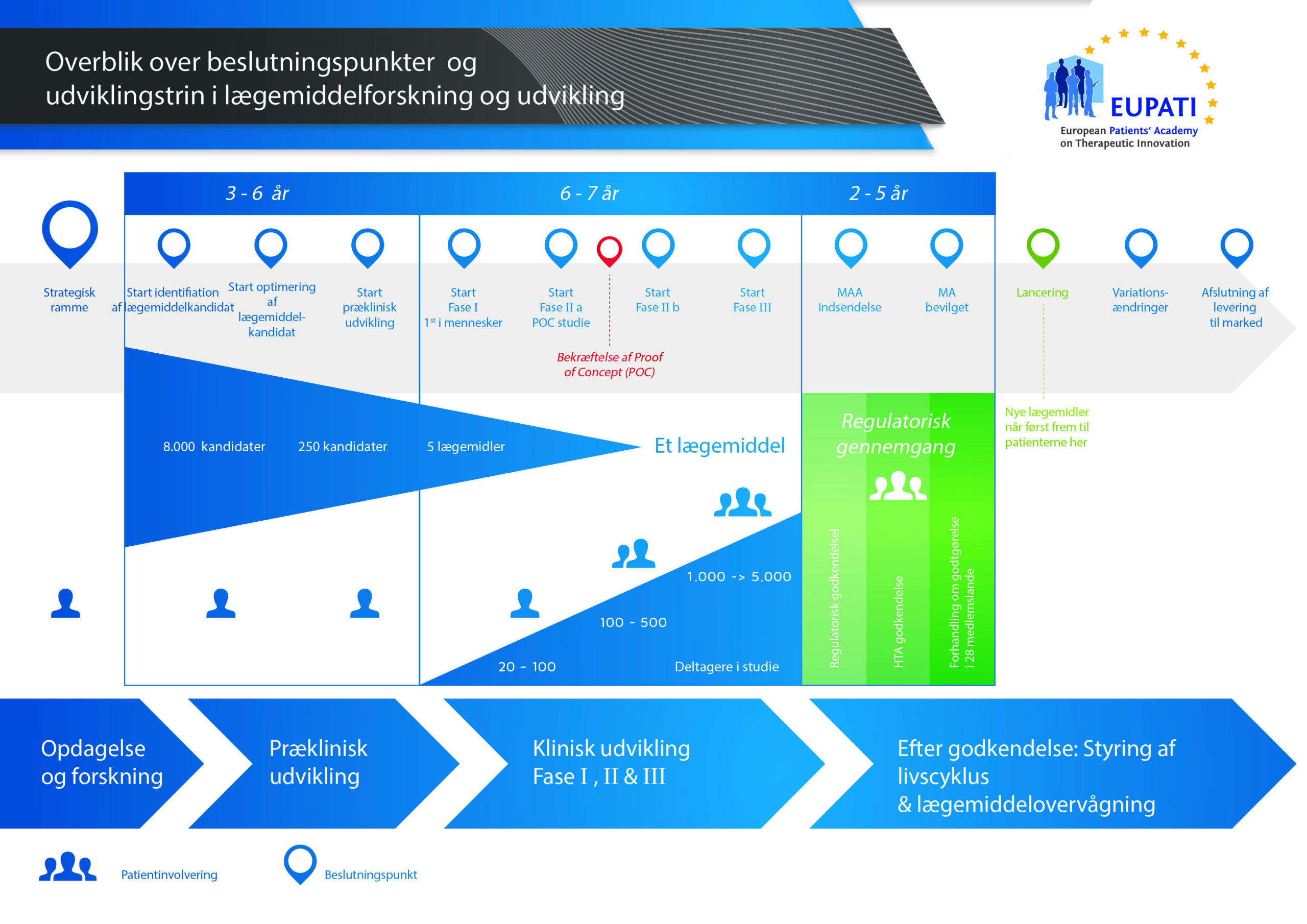
- Det kræver langt over 10 års omhyggelig planlægning og forskning, før et lægemiddel kan tages fra molekyle til behandling, der er klar til at komme på markedet.
Trin 5: Prækliniske sikkerhedstest
Er det sikkert at fortsætte til kliniske test? Denne fase i processen til forskning i lægemidler involverer sikkerhedstest på dyr, som er beskyttet af specifikke regler og bestemmelser i henhold til god laboratoriepraksis (GLP). Et kandidatlægemiddel må ikke testes på mennesker (i kliniske undersøgelser), før dets sikkerhedsprofil er blevet fastlagt i sikkerhedsundersøgelser på dyr. Der er streng kontrol med udvikling af lægemidler. Lovgivningen håndhæver regler og bestemmelser om, hvad der skal gøres, og hvordan det skal gøres.
Før der kan udføres præklinisk arbejde, skal der produceres større mængder af kandidatstoffet, så alle de relevante test kan gennemføres. Denne fremstillingsproces skal også følge strenge retningslinjer og bestemmelser, også kaldet god fremstillingspraksis (GMP).
Disse bestemmelser indeholder oplysninger om, hvilke undersøgelser der skal udføres, og hvilken type dyr der skal bruges til at indhente de nødvendige oplysninger. De omfatter en gennemgang af effekterne:
- i dyret overordnet set
- i alle dyrets væv og organer (systemiske toksicitetsundersøgelser)
- om dyrenes evne til at forplante og udvikle sig normalt (reproduktive toksicitetsundersøgelser)
- på huden eller i øjnene (lokale toksicitetsundersøgelser)
- eventuelle allergier (overfølsomhedsundersøgelser)
- på kromosomerne og generne (genotoksicitetsundersøgelser)
- eventuelle effekter på udvikling af kræft (karcinogenicitetsundersøgelser)
Disse undersøgelser vises nedenfor.
Typer af toksicitetsundersøgelser
- Systematiske toksicitetsundersøgelser
- Undersøgelser af enkeltdosis
- Undersøgelser af gentagen dosis
- Undersøgelser af reproduktionstoksicitet
- Undersøgelser af mandlig fertilitet
- Undersøgelser af kvindelig reproduktion og udvikling
- Lokale toksicitetsundersøgelser
- Overfølsomhedsundersøgelser
- Genotoksicitetsundersøgelser
- Karcinogenicitetsundersøgelser
Disse undersøgelser viser ikke bare sikkerhedsprofilen i dyr, men bidrager også med vigtige oplysninger om:
- hvordan stoffet kommer ind i kroppen (Absorption)
- Distribution af stoffet i kroppen
- nedbrydning af stoffet af kroppen (Metabolisme)
- hvordan stoffet forlader kroppen (Ekskretion)
Det forkortes nogle gange "ADME".
Alle disse oplysninger bruges til at beslutte, om kandidatstoffet kan gå videre til den første (kliniske) undersøgelse på mennesker og – hvis ja – hvilke doser der skal bruges.
For at fortsætte til klinisk test med mennesker skal kandidatstoffet have vist en acceptabel sikkerhedsprofil i alle de nødvendige prækliniske toksicitetsundersøgelser. Det er dog ikke alle de prækliniske sikkerhedsundersøgelser, der vil være fuldførte. Langvarige karcinogenicitetsundersøgelser tager i gennemsnit to år og fortsætter sideløbende med de kliniske forsøg.
Referencer
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford, UK: Wiley-Blackwell.
Bilag
A2-1.02.4-v1.1