Last update: 21 listopada 2016


Dlaczego leki są regulowane?
Każdy chciałby móc otrzymywać leczenie farmaceutyczne, kiedy jest chory. Dlatego potrzebne są leki, które działają skutecznie na chorobę.
Niestety wszystkie leki mają też działania niepożądane. Niezależnie od tego, leki dostępne na rynku muszą być bezpieczne w normalnym użyciu.
Leki muszą być niezawodne. Oznacza to, że muszą być produkowane zgodnie z rygorystycznymi normami jakościowymi.
Wszystkie te wymagania zapewnia się dzięki regulacjom. Leki są podlegają regulacjom, aby mieć pewność, że tylko te, które są wystarczająco bezpieczne, skuteczne i wysokiej jakości, będą wprowadzone do obrotu.
Kto wprowadza regulacje dotyczące leków na poziomie globalnym?
Na poziomie globalnym nie ma żadnych regulacji dotyczących leków. Niemniej przez ponad 20 lat starano się zharmonizować światowe regulacje dotyczące leków. Międzynarodowa Rada Harmonizacji (ICH) zapewnia współpracę pomiędzy organami regulacyjnymi i przemysłem farmaceutycznym w UE, USA, Japonii, Kanadzie, Szwajcarii i innymi organizacjami regionalnymi, ze Światową Organizacją Zdrowia (WHO) oraz licznymi krajowymi organami legislacyjnymi i administracyjnymi, które pełnią rolę obserwatorów.
Kto odpowiada za regulację leków w Unii Europejskiej (UE)?
W UE regulacje dotyczące leków są harmonizowane między wszystkimi państwami członkowskimi na mocy wspólnego zbioru zasad legislacyjnych, a nad procesem czuwają Komisja Europejska (KE), Europejska Agencja Leków (EMA) i krajowe odnośne władze. Zasady te mają także zastosowanie w krajach Europejskiego Obszaru Gospodarczego (EOG), czyli m.in. Norwegii, Islandii i Liechtensteinie. W pozostałej części świata leki są regulowane przez krajowe odnośne władze (ang. National Competent Authority, NCA) każdego kraju i podlegają harmonizacji w regionach ICH.
Ze względu na harmonizację pozwolenie na wprowadzenie leku do obrotu w UE może być wydane jednocześnie w kilku państwach EU i EOG w ramach procedury scentralizowanej, którą nadzoruje Europejska Agencja Leków. Na lek może być także wydane pozwolenie na dopuszczenie do obrotu w państwach członkowskich UE w ramach procedury zdecentralizowanej, procedury wzajemnego uznania i procedury krajowej. Procedury te wiążą się z udziałem krajowych odnośnych władz (ang. National Competent Authority, NCA) i nie mają automatycznie zastosowania do wszystkich państw członkowskich UE.
-
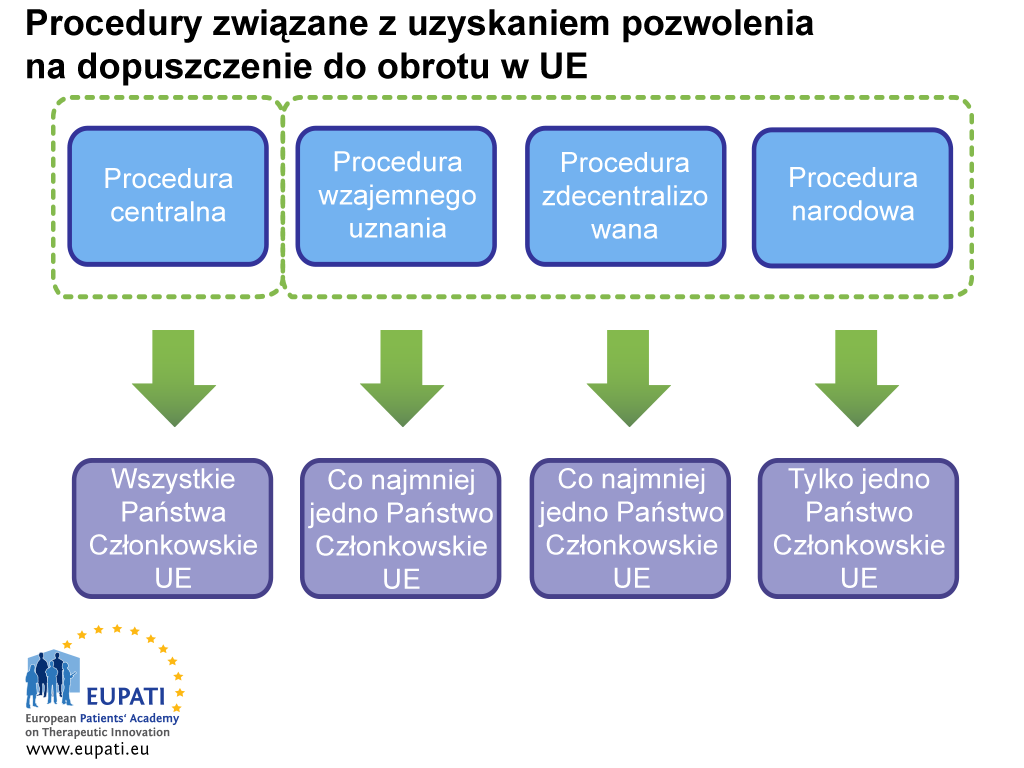
- W uzyskanie pozwolenia na dopuszczenie leku do obrotu zaangażowane są różne strony zależnie od procedury wybranej (lub obowiązującej) sponsora.
Rola Europejskiej Agencji Leków w regulacji leków i wydawaniu pozwoleń
Europejska Agencja Leków (EMA) odgrywa istotną rolę zarówno w regulowaniu leków, jak i wydawaniu na nie pozwoleń.
Europejska Agencja Leków polega na rezultatach badań klinicznych przeprowadzanych przez firmy farmaceutyczne, aby zasięgnąć ich opinii na temat wydania pozwolenia na lek. Zarządza ona także bazą danych badań klinicznych przeprowadzanych na terenie Unii Europejskiej https://www.clinicaltrialsregister.eu/
EMA nadzoruje procedurę scentralizowaną wydawania pozwoleń na dopuszczenie leku do obrotu. Dla większości nowych leków pozwolenia są wydawane w ramach procedury scentralizowanej — wymaga to od firmy włożenia pojedynczego wniosku do EMA, a jeśli lek zostanie zatwierdzony, pozwolenie na dopuszczenie do obrotu we wszystkich państwach UE i EOG tego leku wydaje KE.
Komitetem EMA odpowiedzialnym za ocenę dossier (wniosków) jest Komitet ds. Produktów Leczniczych Stosowanych u Ludzi (ang. Committee for Medicinal Products for Human Use, CHMP). Eksperci oceniający wnioski są wyznaczani przez każde państwo członkowskie i dodatkowo Islandię i Norwegię. Mogą oni korzystać z pomocy maksymalnie pięciu członków współpracujących, wybieranych spośród ekspertów nominowanych przez państwa członkowskie lub EMA i rekrutowanych, jeśli jest taka potrzeba, w celu pozyskania dodatkowych specjalistów z określonej dziedziny naukowej.
A2-5.01.1-V1.1