Last update: 21 ноября 2016


Зачем регулируется обращение лекарственных препаратов?
Каждый хочет иметь возможность получать медицинскую помощь в случае болезни. Поэтому необходимы лекарственные препараты, демонстрирующие эффективность в лечении заболевания.
К сожалению, все лекарственные препараты имеют нежелательные побочные эффекты. Несмотря на это, доступные в продаже лекарственные препараты должны демонстрировать безопасность при нормальном применении.
Лекарственные препараты должны быть надежными. Это означает, что они должны производиться в соответствии со стандартами высокого качества.
Все эти условия рассматриваются в рамках регулирования. Обращение лекарственных препаратов регулируется для обеспечения допуска на рынок только достаточно безопасных, эффективных и высококачественных лекарственных препаратов.
Кто регулирует обращение лекарственных препаратов на международном уровне?
Международного регулирования обращения лекарственных препаратов не существует. Тем не менее, в течение более чем 20 лет прилагаются усилия по гармонизации регулирования обращения лекарственных препаратов на международном уровне. Международный совет по гармонизации (ICH, International Council on Harmonisation) предполагает сотрудничество регуляторных органов и фармацевтической промышленности из ЕС, США, Японии, Канады, Швейцарии, а также других региональных организаций со Всемирной организацией здравоохранения (ВОЗ), а ряд национальных законодательных или административных органов выступают в качестве наблюдателей.
Кто регулирует обращение лекарственных препаратов в Европейском союзе (ЕС)?
В ЕС регулирование обращения лекарственных препаратов гармонизируется среди всех государств-членов на основании общего свода законодательных норм и включает Европейскую комиссию (ЕК), Европейское агентство по лекарственным средствам (EMA, European Medicines Agency) и национальные компетентные органы (NCA, National Competent Authorities) (регуляторные органы). Эти нормы также охватывают Европейскую экономическую зону (ЕЭЗ), к которой относятся Норвегия, Исландия и Лихтенштейн. В остальной части мира обращение лекарственных препаратов регулируется на государственном уровне национальными компетентными органами (NCA) каждой страны и гармонизируется по всем регионам Международного совета по гармонизации (ICH).
Благодаря этой гармонизации лекарственные препараты на территории ЕС могут регистрироваться одновременно во всех странах ЕС и ЕЭЗ при помощи централизованной процедуры, которая осуществляется под надзором Европейского агентства по лекарственным средствам (EMA). Лекарственные препарат также могут быть зарегистрированы в государствах-членах ЕС при помощи децентрализованной процедуры (DCP, Decentralised Procedure), процедуры взаимного признания (MRP, Mutual Recognition Procedure) и национальной процедуры. В этом процессе задействованы национальные компетентные органы (NCA) и он не применяется автоматически ко всем Европейским государствам-членам.
-
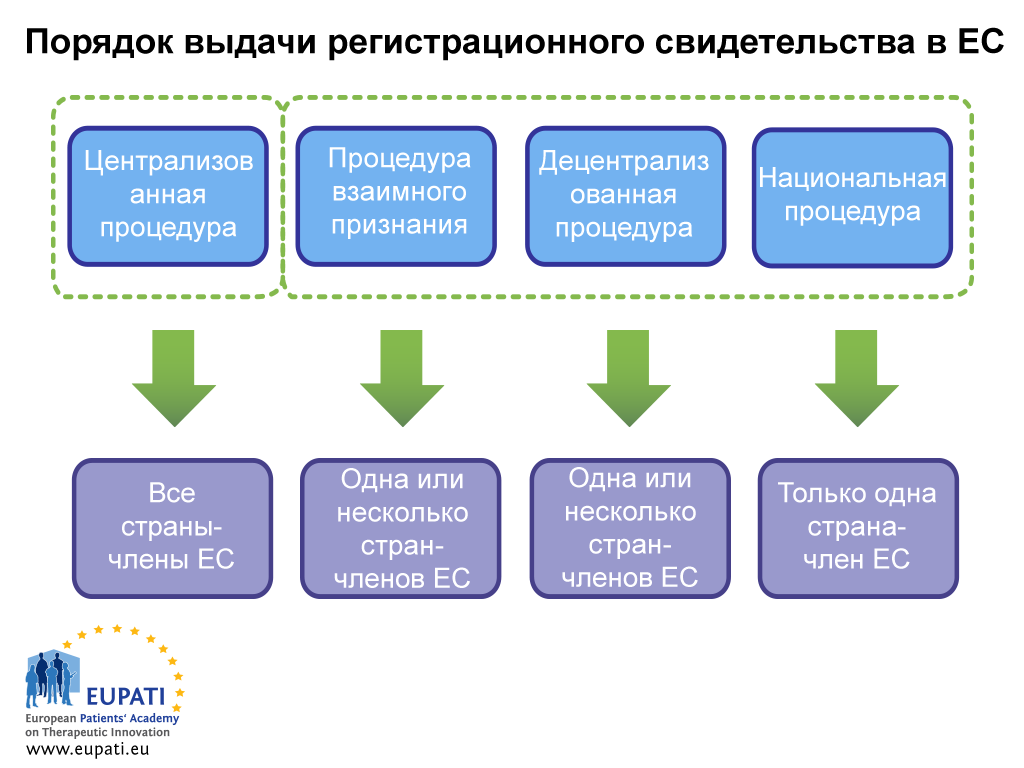
- зависимости от того, какую процедуру выбирает спонсор (или какой он вынужден следовать), процесс получения регистрационного свидетельства медицинского препарата может требовать участия разных сторон.
Роль Европейского агентства по лекарственным средствам в регулировании обращения лекарственных препаратов и их регистрации
Европейское агентство по лекарственным средствам (EMA) играет важную роль в регулировании обращения лекарственных препаратов и их регистрации.
Европейское агентство по лекарственным средствам опирается на результаты клинических исследований, проведенных фармацевтическими компаниями, чтобы прийти к выводу в отношении регистрации лекарственных препаратов. Агентство также ведет базу данных клинических исследований, которые проводятся в Европейском союзе https://www.clinicaltrialsregister.eu/
Европейское агентство по лекарственным средствам (EMA) руководит централизованной процедурой (CP, Centralised Procedure) для получения регистрационного свидетельства (РС). Большинство новых лекарственных препаратов регистрируются при помощи централизованной процедуры. Она требует, чтобы компания подала единую заявку в Европейское агентство по лекарственным средствам (EMA) и, в случае успешной регистрации выдается лицензия ЕК на сбыт лекарственного препарата во всех странах ЕС и ЕЭЗ.
Комитетом Европейского агентства по лекарственным средствам (EMA), который несет ответственность за оценку досье (заявок), является Комитет по лекарственным препаратам для медицинского применения (CHMP). Эксперты, которые оценивают заявки на регистрацию, выдвигаются каждым государством-членом и дополнительно Исландией и Норвегией. Они могут поддерживаться не более чем пятью кооптированными членами, выбранными среди экспертов, выдвинутых государствами-членами или Европейским агентством по лекарственным средствам (EMA) и нанятыми, при необходимости, для обеспечения дополнительной экспертизы в той или иной научной области.
A2-5.01.1-V1.1