

Why are medicines regulated?
Everybody wants to be able to receive medical treatment if they are ill. Therefore medicines are needed that are effective against the illness.
Unfortunately all medicines come with unwanted side effects. Irrespective, medicines on the market must be safe for normal use.
Medicines must be reliable. This means that they must be manufactured according to high quality standards.
All these points are addressed through regulation. Medicines are regulated to ensure that only sufficiently safe, effective and high quality medicines can be marketed.
Who regulates medicines globally?
There is no global regulation of medicines. However for more than 20 years, efforts have been ongoing to harmonise medicines regulation globally. The International Council on Harmonisation (ICH) involves collaboration between regulatory authorities and the pharmaceutical industry from the EU, USA, Japan, Canada, Switzerland, and other regional organisations, with the Word Health Organisation (WHO) and a number of national legislative or administrative authorities acting as observers.
Who regulates medicines in the European Union (EU)?
In the EU, medicines regulation is harmonised across all member states under a common set of legislative rules, and involves the European Commission (EC), the European Medicines Agency (EMA) and the National Competent Authorities (NCAs) (regulatory authorities). These rules also cover the European Economic Area (EEA), which includes Norway, Iceland, and Liechtenstein. In the rest of the world medicines are regulated at the national level by each country’s National Competent Authority (NCA), and harmonised across the ICH regions.
Due to this harmonisation, medicines in the EU may be authorised simultaneously in all the countries of the EU and EEA through the Centralised Procedure, which is overseen by the EMA. A medicine can also be authorised in EU Member States through the Decentralised Procedure (DCP), the Mutual Recognition Procedure (MRP), and the National Procedure. These involve national competent authorities (NCAs) and do not automatically apply to all European Member States.
-
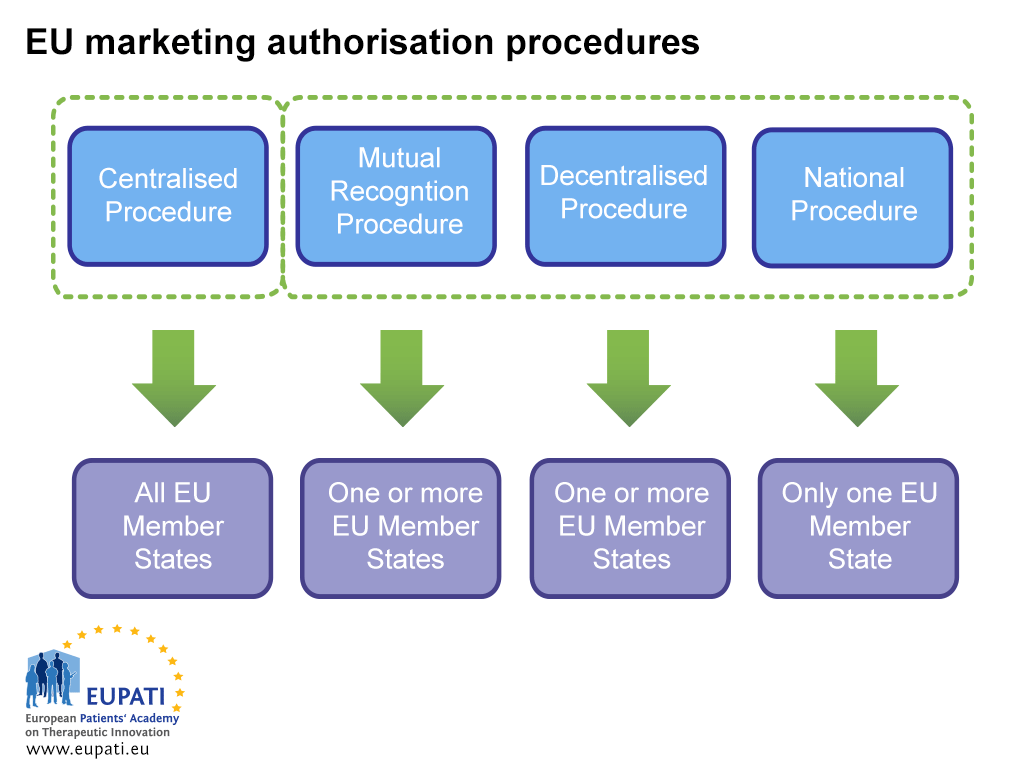
- Different actors are involved in the marketing authorisation of a medicine depending on which procedure the sponsor opts (or is obliged) to follow.
The role of the European Medicines Agency in medicines regulation and approval
The European Medicines Agency (EMA) plays an important role both in medicines regulation and medicines approval.
The European Medicines Agency relies on the results of clinical trials carried out by pharmaceutical companies to reach its opinions on the authorisation of medicines. It also manages a database of clinical trials carried out in the European Union https://www.clinicaltrialsregister.eu/
The EMA oversees the Centralised Procedure (CP) for Marketing Authorisation (MA). Most new medicines seek authorisation through the CP – it requires the company to submit a single application to the EMA, and if the medicine is approved then a license is granted by the EC for the medicine to be marketed in all EU and EEA countries.
The EMA committee responsible for assessing the dossiers (applications) is the Committee for Medicinal Products for Human Use (CHMP). The experts that assess the submissions are nominated by each of the Member States and additionally Iceland and Norway. They can be supported by up to five co-opted members, chosen among experts nominated by Member States or the EMA and recruited, when necessary, to provide additional expertise in a particular scientific area.
-
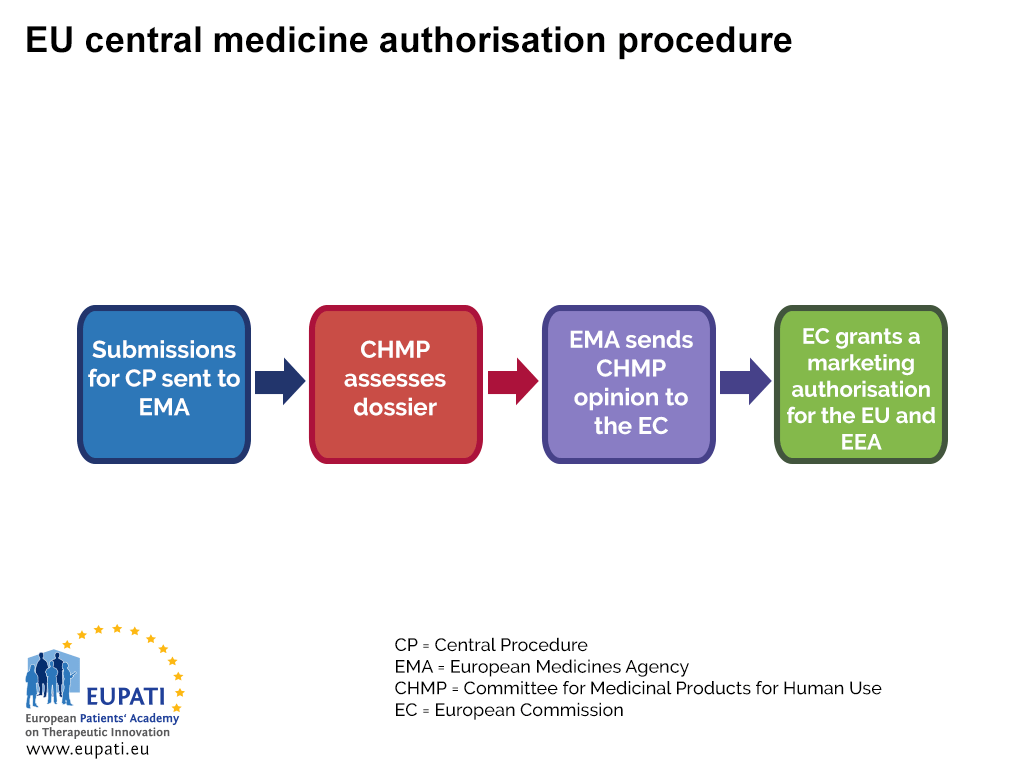
- The centralised procedure used to authorise a medicine in all EU and EEA countries.
A2-5.01.1-V1.1