Last update: 3 August 2015


Einleitung
Es dauert über 12 Jahre und kostet durchschnittlich mehr als eine Milliarde Euro, all die Forschungs- und Entwicklungsarbeiten durchzuführen, die erforderlich sind, bis ein neues Arzneimittel für die Behandlung von Patienten zur Verfügung steht.
Arzneimittelentwicklung ist ein risikoreiches Geschäft. Der größte Teil (ca. 98 %) neu entwickelter Wirkstoffe schafft es nicht, als neues Arzneimittel auf den Markt zu gelangen. Das liegt daran, dass das Verhältnis zwischen dem Nutzen und den im Verlauf der Entwicklung festgestellten Risiken (schädliche Nebenwirkungen) dem Vergleich mit anderen, bereits für die Behandlung von Patienten verfügbaren Arzneimitteln meist nicht standhält.
Die Entwicklung eines neuen Arzneimittels kann in zehn unterschiedliche Schritte unterteilt werden. Der folgende Artikel behandelt Schritt 9: Behördliche Überprüfung und Antrag auf Marktzulassung.
-
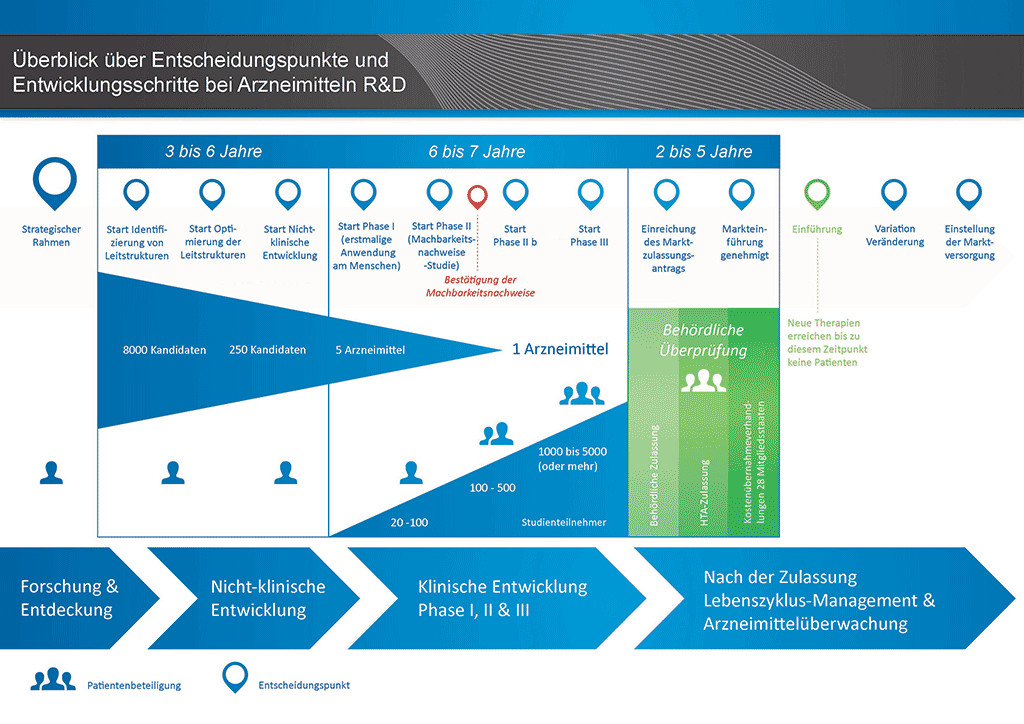
- Es benötigt mehr als 10 Jahre sorgfältiger Planung und Forschung, bis ein Arzneimittel sich vom Molekül zur marktfähigen Behandlung entwickelt hat.
Schritt 9: Behördliche Überprüfung (Antrag auf Marktzulassung)
Zeigen die Ergebnisse der klinischen Phase-III-Studien ein akzeptables Nutzen-Risiko-Verhältnis, kann ein Antrag auf Marktzulassung erstellt werden. Sämtliche Informationen (nicht-klinisch, klinisch und herstellungsbezogen) werden gesammelt und in einem vorgegebenen Format zusammengestellt. Dieser nennt sich „Dossier“ und wird bei den Zulassungsbehörden eingereicht. Die Erfahrung der Mitarbeiter in den Zulassungsabteilungen ist sehr wichtig, da die verschiedenen Zulassungsbehörden in aller Welt leicht abweichende Anforderungen haben.
Die Internationale Konferenz zur Harmonisierung technischer Anforderungen für die Zulassung von Humanarzneimitteln (ICH) harmonisierte viele Anforderungen für die USA, Europa und Japan. Dies führte dazu, dass Untersuchungen nicht mehr mehrfach durchgeführt werden müssen, und vereinfachte den Prozess der Erstellung des einzureichenden Common Technical Document (standardisierter Antrag auf Zulassung eines Arzneimittels).
Nach dem Erhalt des Dossiers prüft die Zulassungsbehörde die Informationen; die Beantwortung etwaiger Rückfragen der Behörde ist Sache der Mitarbeiter der Zulassungsabteilung, die das Dokument eingereicht hat. Sobald die Zulassungsbehörde mit den Ergebnissen (Risiko/Nutzen-Beurteilung) zufrieden ist, wird sie ihre Zustimmung zur Vermarktung des neuen Arzneimittels geben. Der Prüfprozess dauert in der Regel 12 bis 18 Monate. Dieser Zeitraum kann in speziellen Fällen in Abstimmung mit der Zulassungsbehörde verkürzt werden. Stellen sich viele Fragen, kann er aber auch länger dauern. Die Behörden können ggf. auch weitere klinische Studien verlangen, bevor sie ihre Zustimmung geben. Das Arzneimittel wird für die Vermarktung erst freigegeben, wenn die Zulassungsbehörden überzeugt sind. In manchen Fällen liegen Umstände vor, die von den Zulassungsbehörden nicht akzeptiert werden können, und das Arzneimittel erhält keine Erlaubnis zur Markteinführung.
In vielen Ländern werden zudem Untersuchungen zur Wirtschaftlichkeit des neuen Arzneimittels verlangt. Diese Dokumente werden der Regierung und Kostenträgern dabei helfen, auf Grundlage einer Gesundheitstechnologiefolgenabschätzung (Health Technology Assessment, HTA) eine Entscheidung hinsichtlich der Verordnungs- und Erstattungsfähigkeit (d. h. der Übernahme der Kosten des Arzneimittels durch Krankenkasse/Krankenversicherung) zu treffen und entsprechende Empfehlungen auszuarbeiten.
Eine bekannte Institution, die sich mit der Gesundheitstechnologiefolgenabschätzung befasst, ist das Nationale Institut für klinische Exzellenz (National Institute for Clinical Excellence, NICE) im Vereinigten Königreich (UK). NICE spricht Empfehlungen aus, ob die Regierung die Verschreibung des Arzneimittels erlauben soll.
Quellenangaben
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford: Wiley-Blackwell.
Anlagen
- Präsentation: Die grundlegenden Prinzipien der Arzneimittelentdeckung und -entwicklung
Size: 945,895 bytes, Format: .pptx
Die grundlegenden Prinzipien der Arzneimittelentdeckung und -entwicklung. Es dauert über 12 Jahre und kostet mehr als eine Milliarde Euro, all die Forschungs- und Entwicklungsarbeiten durchzuführen, die erforderlich sind, bis ein neues Arzneimittel für die Behandlung von Patienten zur Verfügung steht. Diese Präsentation stellt die Details des Prozesses von der Entdeckung bis zur Markteinführung eines neuen Arzneimittels und darüber hinaus vor.
A2-1.02.8-v1.1