Last update: 3 augustus 2015


Inleiding
Gemiddeld duurt het ruim 12 jaar en kost het meer dan 1 miljard euro om al het benodigde onderzoeks- en ontwikkelingswerk te doen voordat een nieuw geneesmiddel beschikbaar komt om door patiënten te worden gebruikt.
Geneesmiddelenontwikkeling is een risicovolle onderneming. De meeste stoffen (ongeveer 98%) die worden ontwikkeld, halen de markt niet als nieuw geneesmiddel. Dit komt voornamelijk doordat de voordelen en risico’s (negatieve bijwerkingen) die tijdens de ontwikkeling worden geconstateerd, zich slecht verhouden tot geneesmiddelen die al verkrijgbaar zijn voor patiënten.
De ontwikkeling van een nieuw geneesmiddel kan worden onderverdeeld in 10 verschillende stappen. Het volgende artikel gaat over Stap 9: Indiening en aanvraag voor een handelsvergunning bij de toezichthoudende instanties.
-
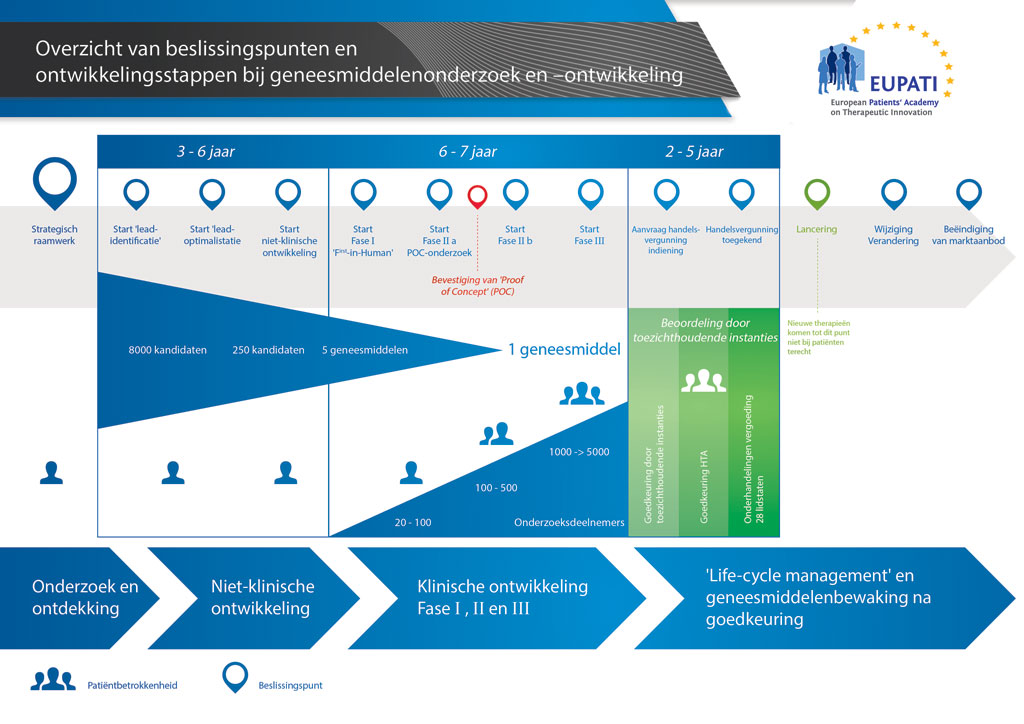
- Er kunnen meer dan 10 jaar aan nauwkeurig plannen en onderzoek nodig zijn om een geneesmiddel te ontwikkelen van een molecuul tot een verkoopbare behandeling.
Stap 9: Indiening bij de toezichthoudende instanties (vergunningaanvraag)
Als de resultaten van de klinische onderzoeken in fase III duiden op een aanvaardbare baten-risicorelatie, kan een aanvraag voor een handelsvergunning worden opgesteld. Alle informatie (niet-klinisch, klinisch en over de vervaardiging) wordt verzameld en geordend volgens een vooraf vastgestelde structuur. Dit wordt aangeduid als een ‘dossier’ en naar de toezichthoudende instanties gestuurd. Deskundigheid van de medewerkers op de registratieafdeling van de farmaceutische bedrijven is onontbeerlijk, omdat er kleine verschillen kunnen zijn in de vereisten van de uiteenlopende toezichthoudende instanties over de wereld.
De International Committee on Harmonisation (ICH) harmoniseerde veel vereisten voor de VS, Europa en Japan. Dit heeft het aantal dubbele proeven verminderd en de procedure vereenvoudigd, wat leidde tot een Common Technical Document (CTD) voor beoordeling.
Zodra het dossier is ontvangen, beoordeelt de toezichthoudende instantie de informatie en worden er vragen voorgelegd aan de medewerkers van de registratieafdeling die het document heeft opgestuurd. Als de toezichthoudende instantie tevreden is met de resultaten (baten-risicobeoordeling) wordt goedkeuring verleend om het nieuwe geneesmiddel in de handel te brengen. De beoordelingsprocedure duurt meestal 12-18 maanden. Deze periode kan korter zijn in speciale gevallen die door de toezichthoudende instanties zijn overeengekomen, maar kan worden verlengd als er te veel vragen open blijven. De autoriteiten kunnen meer klinische onderzoeken eisen voordat ze zich bereid verklaren om goedkeuring te verlenen. Het geneesmiddel mag niet in de handel worden gebracht voordat de toezichthoudende instanties tevreden zijn. Soms zijn er omstandigheden die niet door de toezichthoudende instanties kunnen worden aanvaard, en wordt er geen toestemming verleend om het geneesmiddel in de handel te brengen.
In veel landen zijn ook onderzoeken naar de kosten-effectiviteit van het nieuwe geneesmiddel vereist. Deze documenten helpen de overheden of verzekeraars via groepen voor Health Technology Assessment om te besluiten, en adviseren of het geneesmiddel mag worden voorgeschreven en betaald via het verzekeringsstelsel in het land.
Een bekende HTA-instantie is het National Institute for Clinical Excellence (NICE) in Groot-Brittannië. NICE adviseert of de overheid wel of niet moet toestaan dat het geneesmiddel mag worden voorgeschreven.
Referenties
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford: Wiley-Blackwell.
Bijlagen
- Factsheet: Indiening bij de toezichthoudende instanties
Size: 103,601 bytes, Format: .docx
Dit factsheet geeft informatie over de activiteiten rondom de indiening van het uiteindelijke dossier over het geneesmiddel bij de registratieautoriteiten in voorbereiding op de mogelijke introductie ervan op de markt.
- Presentatie: De basisprincipes van geneesmiddelontdekking en -ontwikkeling
Size: 950,426 bytes, Format: .pptx
De basisprincipes van geneesmiddelontdekking en -ontwikkeling. Gemiddeld duurt het ruim 12 jaar en kost het meer dan 1 miljard euro om al het benodigde onderzoeks- en ontwikkelingswerk te doen voordat een nieuw geneesmiddel beschikbaar komt om door patiënten te worden gebruikt. Deze presentatie gaat nader in op het proces van ontdekking tot marktintroductie van een nieuw geneesmiddel en daarna.
A2-1.02.8-v1.1