

Introduction
It takes over 12 years and on average costs over €1 billion to do all the research and development necessary before a new medicine is available for patients to use.
Medicines development is a high-risk venture. The majority of substances (around 98%) being developed do not make it to the market as new medicines. This is mostly because when you look at the benefits and risks (negative side effects) found during development do not compare well with medicines that are already available to patients.
The development of a new medicine can be divided into 10 different steps. The following article covers Step 9: Regulatory submission and marketing authority authorisation application.
-
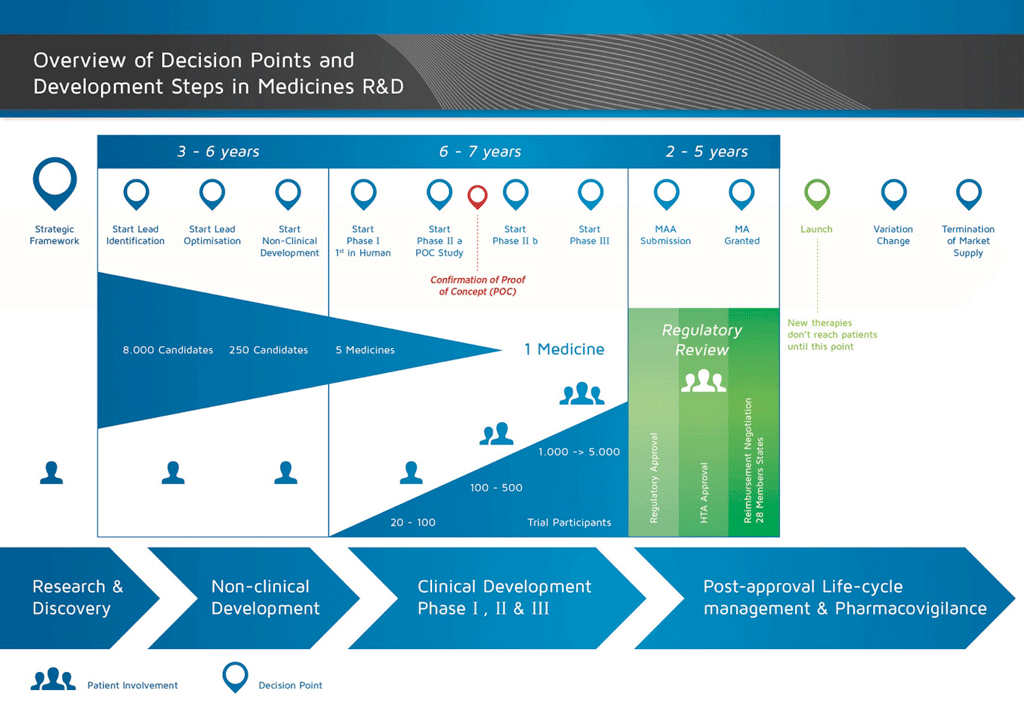
- It takes well over 10 years of careful planning and research for a medicine to go from molecule to a marketable treatment.
Step 9: Regulatory submission (Marketing authorisation application)
If the results of the Phase III clinical studies show an acceptable benefit-risk relationship, a Marketing Authorisation Application (MAA) can be prepared. All the information (non-clinical, clinical, and manufacturing) is collected and organised in a pre-determined format. This is called a ‘dossier’ and it is sent to the regulatory authorities. The expertise of the staff in the regulatory departments is very important as the different regulatory authorities around the world have slightly different requirements.
The International Committee on Harmonisation (ICH) harmonised many of the requirements for the USA, Europe, and Japan. This has reduced duplication of testing and has simplified the process resulting in a Common Technical Document (CTD) for review.
Once the dossier is received, the regulatory authority will review the information and submit questions to be answered by the staff in the regulatory department who sent the document. Once the regulatory authority is satisfied with the results (risk-benefit assessment) they will give their approval for the new medicine to be marketed. The review process usually takes 12-18 months. This period can be shorter in special cases agreed by the regulatory authorities, but can be prolonged if there are many questions to answer. The authorities may require more clinical studies before they are prepared to give their approval. The medicine will not be allowed to enter the market until the regulatory authorities are satisfied. Sometimes there are conditions that cannot be accepted by the regulatory authorities, and the medicine will not be accepted to go into the market.
In many countries, studies on the cost effectiveness of the new medicine are also required. These documents will support the government or insurance companies through Health Technology Assessment (HTA) groups to decide and give recommendations about allowing the medicine to be prescribed and paid by the insurance system in the country.
One well-known HTA body is the National Institute for Clinical Excellence (NICE) in the United Kingdom. NICE recommends whether or not the government should allow the medicine to be prescribed.
References
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford: Wiley-Blackwell.
Attachments
- Fact sheet: Regulatory submission
Size: 103,601 bytes, Format: .docx
This fact sheet details the activities surrounding the submission of the final dossier on the medicine to the regulative authorities in preparation for its potential release on to the market.
- Presentation: The basic principles of medicine discovery and development
Size: 918,164 bytes, Format: .pptx
The basic principles of medicine discovery and development. It takes over 12 years and over €1 billion to do all the research and development necessary before a new medicine is available for patients to use. This presentation details the process from discovery to release of a new medicine onto the market and beyond.
A2-1.02.8-v1.1