Last update: 3 августа 2015


Введение
В среднем на все исследования и разработки, необходимые для того, чтобы новый лекарственный препарат был доступен для пациентов, уходит более 12 лет и более 1 миллиарда евро.
Разработка лекарственных препаратов — это рисковый бизнес. Большинство разрабатываемых соединений (около 98 %) так и не выходят на рынок. Так происходит потому, что при оценке преимуществ и рисков (негативных побочных эффектов), обнаруживаемых в ходе разработки, сложно сравнивать их с уже имеющимися на рынке препаратами.
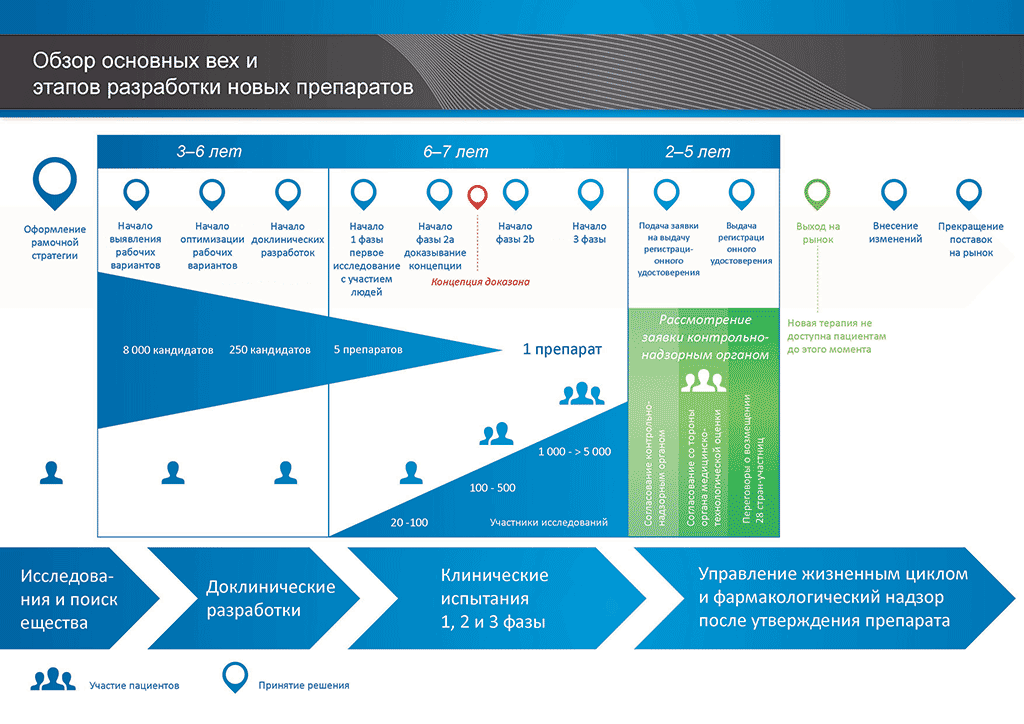
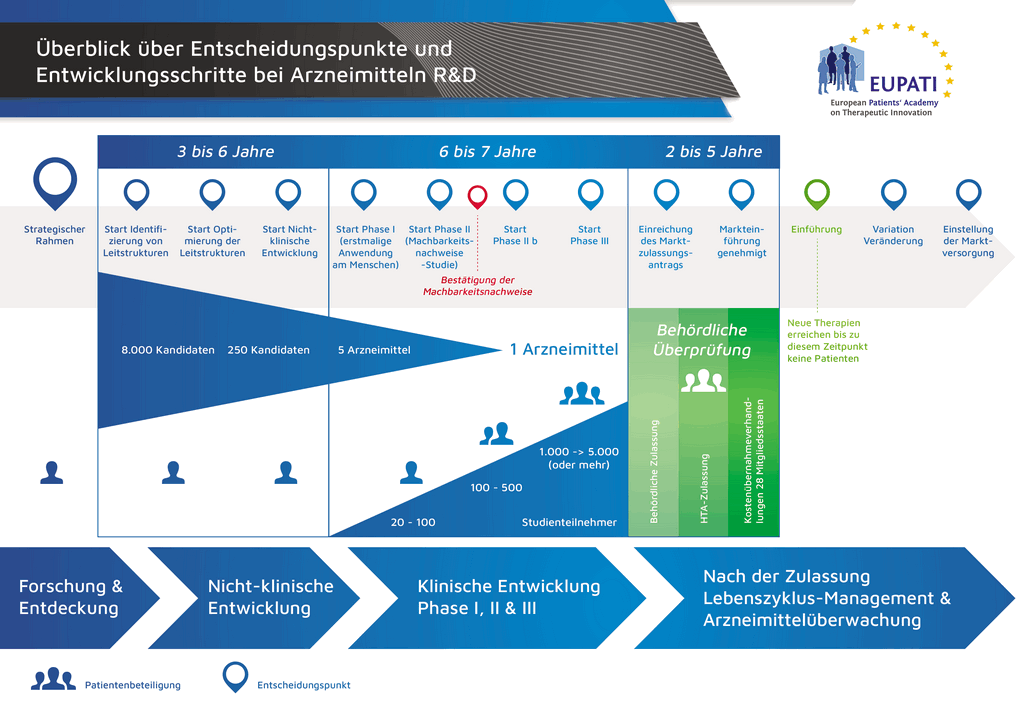
Процесс разработки нового лекарственного препарата можно представить в 10 шагах. В следующей статье описывается 9-й шаг. Подача заявки в контрольно-надзорные органы и заявки на выдачу регистрационного свидетельства
-
- С момента создания молекулы до момента начала продажи медицинского препарата проходит больше 10 лет, необходимых для тщательнейшего планирования и исследований.
-
- Es benötigt mehr als 10 Jahre sorgfältiger Planung und Forschung, bis ein Arzneimittel sich vom Molekül zur marktfähigen Behandlung entwickelt hat.
Шаг 9: Подача заявки в контрольно-надзорные органы (на выдачу регистрационного свидетельства)
Если по результатам клинических исследований 3 фазы установлено допустимое соотношение преимуществ и рисков, может быть оформлена заявка на выдачу регистрационного свидетельства (Marketing Authorisation Application, МАА). Производится сбор и классификация всех данных (доклинических, клинических и производственных) согласно установленному формату. Такой документ называется «досье», которое направляют в контрольно-надзорные органы. Квалификация специалистов контрольно-надзорных органов чрезвычайно важна, поскольку требования таких органов в разных странах мира могут несколько различаться.
Международный комитет по гармонизации (ICH) обеспечил унификацию многих требований в США, Европе и Японии. Это позволило снизить количество дублирующих друг друга испытаний и упростило процесс, который теперь позволяет подготавливать «общий технический документ» (CTD) на рассмотрение.
После получения досье контрольно-надзорный орган рассматривает данные и направляет вопросы специалистам, направившим документ. Когда контрольно-надзорный орган счел результаты (соотношение преимуществ и рисков) удовлетворительными, он выдает разрешение на коммерческий выпуск нового препарата. Процесс рассмотрения обычно занимает 12–18 месяцев. Этот срок может быть сокращен в отдельных случаях, согласованных с контрольно-надзорными органами, но может быть и более продолжительным, если необходимо проработать много вопросов. До выдачи разрешения контрольными органами последние могут потребовать проведения дополнительных клинических исследований. Выход лекарственного препарата на рынок невозможен, если он не удовлетворяет требованиям контрольно-надзорных органов. Некоторые параметры могут быть неприемлемыми для согласования контрольно-надзорными органами, и тогда препарат не выходит на рынок.
Во многих странах также необходимо проведение исследований об экономической эффективности нового препарата. Такие документы помогают госорганам и страховым компаниям, через группы «оценки медицинских технологий» (HTA), принять решение, разрешить ли препарат к назначению врачами и оплачивать ли его через страховую систему, установленную в этой стране.
Один из известных органов по оценке медицинских технологий — Национальный институт здоровья и качества медицинской помощи Великобритании (National Institute for Clinical Excellence, NICE). NICE выдает государству рекомендации насчет утверждения препаратов.
Справочная литература
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford: Wiley-Blackwell.
Приложения
- Информационный бюллетень: Подача заявки в контрольно-надзорный орган
Size: 97,801 bytes, Format: .docx
В этом информационном бюллетене приводится порядок действий при подаче окончательного досье на лекарственный препарат в контрольно-надзорные органы в ходе подготовки его возможного выпуска на рынок.
A2-1.02.8-v1.1