Last update: 5 Ottobre 2016


Introduzione
Il processo di sviluppo dei farmaci comporta un lungo percorso. L’obbiettivo ultimo di qualsiasi processo di sviluppo è l’approvazione per l’immissione in commercio del nuovo prodotto medicinale, un’autorizzazione all’immissione in commercio (Marketing Authorisation, MA). Entro le aziende farmaceutiche, i dipartimenti che si occupano degli affari normativi (Regulatory Affairs, RA) sono parte integrante di tutte le fasi nel corso del ciclo di vita del prodotto medicinale. Il dipartimento degli affari normativi è responsabile in particolare per le richieste che devono essere presentate prima di ogni studio clinico, la preparazione e la presentazione del dossier per la richiesta di autorizzazione all’immissione in commercio (Marketing Authorisation Application, MAA), e altre attività successive alla concessione della MA, ad esempio una richiesta di modifica all’autorizzazione all’immissione in commercio (una variazione). I professionisti degli affari normativi hanno bisogno di possedere una conoscenza esauriente di tutti i regolamenti relativi ai farmaci e dell’intero processo di sviluppo.
-
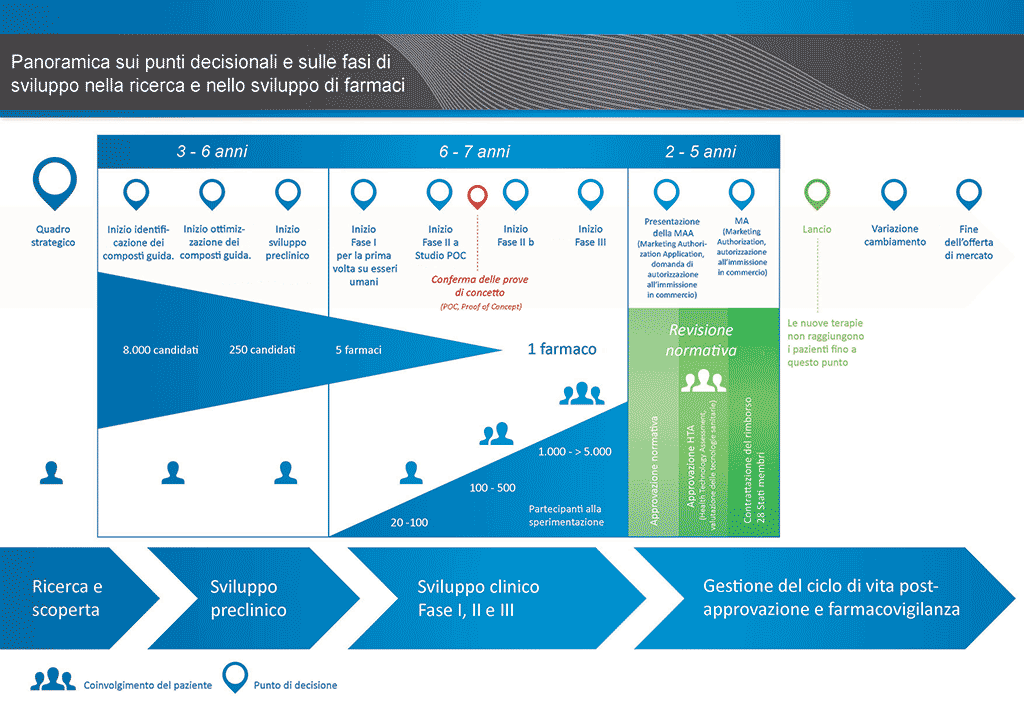
- Occorrono oltre 10 anni di attenta pianificazione e ricerca perché un farmaco passi da molecola a trattamento disponibile sul mercato.
Figura 1– Resoconto del processo di sviluppo dei farmaci
Presentazioni per l'autorizzazione all'immissione in commercio (Marketing Authorisation, MA)
L'azienda farmaceutica deve decidere in uno stadio iniziale di sviluppo quale tipo di richieste presentare per l'autorizzazione all'immissione in commercio, ad es.:
- Una domanda completa: vedere il triangolo relativo al Documento tecnico comune (Common Technical Document, CTD) sotto.
- Una domanda abbreviata (domanda ridotta).
- Una domanda bibliografica, basata sulla letteratura scientifica esistente.
Le richieste necessitano della presentazione di un dossier con le documentazione per gli enti competenti. La figura 1 illustra il processo di sviluppo per un farmaco nuovo e innovativo. Questo tipo di farmaco richiede la presentazione di un dossier completo, in cui devono essere inclusi tutti gli elementi della documentazione per il farmaco.
Quali elementi include un dossier?
La figura 2 mostra gli elementi che costituiscono il documento tecnico comune (Common Technical Document, CTD), il dossier presentato agli enti normativi come richiesta di autorizzazione all'immissione in commercio (MAA) in Canada, Europa, Giappone, Svizzera, Stati Uniti e altri. Il formato del CTD è stato sviluppato dal Consiglio internazionale sull'armonizzazione (International Council for Harmonisation, ICH) dei requisiti tecnici per i prodotti farmaceutici per uso umano. Il CTD deve essere utilizzato per tutti i tipi di MAA nell'UE indipendentemente dalla procedura (procedura centralizzata, CP, procedura di mutuo riconoscimento, MRP, procedura centralizzata, DCP, oppure procedura nazionale, NP) o il tipo di richiesta (autonoma, generici ecc.). Il formato del CTD è applicabile per tutti i tipi di prodotto (nuove sostanze chimiche, radiofarmaceutici, vaccini, prodotti erboristici ecc.).
Nel CTD sono presenti cinque moduli distinti.
- Modulo 1: Informazioni amministrative regionali.
- Modulo 2: Sommari e resoconti.
- Modulo 3: Qualità.
- Modulo 4: Relazioni su studi non clinici.
- Modulo 5: Relazioni sugli studi clinici.
I moduli del CTD dal 2 al 5 sono comuni per tutte le regioni, mentre il Modulo 1 è specifico per ciascuna regione e non è considerato parte del CTD stesso. Nei moduli dal 3 al 5 si trova la maggior parte della documentazione relativa alla qualità, sicurezza ed efficacia del farmaco. I professionisti che si occupano di affari normativi garantiscono che la documentazione presentata sia in linea con tutti i regolamenti, le direttive e le linee guida pertinenti. Questi ultimi producono anche i sommari inclusi nel Modulo 2.
Figura 2: Il triangolo relativo al documento tecnico comune (CDT).
Modulo 1: Informazioni amministrative regionali
Il modulo 1 del CTD contiene tutte le informazioni amministrative necessarie a livello regionale. L'Unione Europea dispone della propria versione del modulo 1, costituita dai seguenti 10 elementi:
1.0 Lettera di accompagnamento
1.1 Indice completo
1.2 Modulo di richiesta
1.3 Informazioni sul prodotto
Questa è un'informazione che sarà usata sia dai professionisti sanitari che dai pazienti. Include il Riassunto delle caratteristiche del prodotto (RCP), un documento dettagliato per i professionisti sanitari, l'etichettatura e il foglio illustrativo (FI). Per dimostrare che il FI sia comprensibile dal pubblico generale, deve essere effettuato un test di leggibilità. Le autorità nazionali competenti e l'EMA dispongono di moduli pubblicati in tutte le lingue dell'UE per la presentazione delle informazioni relative al prodotto le quali prescrivono in dettaglio il formato e il contenuto.
1.4 Informazioni sugli esperti
Il Modulo 2 del CTD contiene i sommari e i resoconti scritti da esperti. Ciascuno di questi esperti deve fornire un curriculum vitae (CV) e firmare una dichiarazione che hanno seguito le regole di tutti i regolamenti e le direttive pertinenti nella creazione dei sommari.
1.5 Requisiti specifici per diversi tipi di richieste
Informazioni aggiuntive necessarie per tipi particolari di richieste, come domande bibliografiche, generici, richieste per "ibridi" o biosimilari, dati (estesi) / esclusività di mercato, domanda in circostanze eccezionali o richiesta per immissione in commercio condizionale
1.6 Valutazione ambientale dei rischi
Tutti i principi attivi presenti nei farmaci possono porre un potenziale rischio per l'ambiente, e tutte le sostanze e i loro metaboliti in conclusione finiranno nell'ambiente. L'azienda deve affrontare il possibile impatto ambientale creato dall'uso, stoccaggio e smaltimento del farmaco.
1.7 Informazioni relative all'esclusività del mercato dei farmaci orfani
Se il farmaco è progettato come farmaco orfano con lo scopo di trattare una malattia rara, sono necessarie informazioni particolari. Se un altro farmaco sul mercato ha già l'esclusività del mercato per la stessa indicazione, il nuovo farmaco può essere approvato solo in particolari condizioni.
1.8 Informazioni relative alla farmacovigilanza
Deve essere inclusa una descrizione della farmacovigilanza e dei sistemi di gestione del rischio. Il richiedente deve dimostrare che vi sia un'adeguata sorveglianza sulle reazioni avverse e sui rischi potenziali. Deve comprendere la prova che il richiedente dispone di una persona qualificata responsabile per la farmacovigilanza e dei mezzi necessari per la notifica di qualsiasi reazione avversa che si verifichi nell'UE o in un paese terzo (Articolo 8 (n) della Direttiva 2001/83/CE).
1.9 Informazioni relative agi studi clinici
Il richiedente deve includere una dichiarazione che qualsiasi studio clinico del farmaco eseguito fuori dell'UE soddisfa i regolamenti dell'UE.
1.10 Informazioni relative agli studi pediatrici
Nell'UE, tutti i nuovi farmaci devono essere considerati per la popolazione pediatrica. In generale, devono essere studiati su bambini. Tuttavia, può essere concessa una deroga a tale requisito se la malattia viene osservata solo in individui anziani o adulti, oppure se è probabile che il nuovo farmaco sia inefficace o non sicuro in parte o in tutta la popolazione pediatrica. Se non viene concessa una deroga, l'azienda deve produrre un Piano di sperimentazione pediatrica (Paediatric Investigation Plan, PIP), a meno che non sia stato concesso un rinvio, nel cui caso il PIP può essere prodotto successivamente. Tale sezione deve comprendere una copia della deroga o della decisione relativa al PIP (tra cui i rinvii, se applicabile).
Com'è organizzato il dossier?
Nella maggioranza dei casi, le presentazioni in cartaceo non sono più possibili, cioè tutta la documentazione nei cinque moduli del CTD deve essere redatta in formato elettronico standard: l' eCTD. L' eCTD non è solo una raccolta di documenti in PDF, né un enorme singolo file in PDF. Piuttosto, l'eCTD è uno standard che descrive in dettaglio la cartella e la struttura del file che deve essere seguita in modo che l'azienda e gli enti di regolamentazione possano navigare facilmente nel dossier, come si fa solitamente in una normale directory di computer.
Che cos'è il processo di presentazione?
Il richiedente deve prendere attentamente in considerazione tutti i problemi logistici e normativi prima della presentazione. Ciò include la scelta della procedura di autorizzazione all'immissione in commercio da perseguire: la procedura centralizzata (Centralised Procedure, CP), la procedura di mutuo riconoscimento (Mutual Recognition Procedure, MRP) o la procedura decentralizzata (Decentralised Procedure, DCP), oppure la procedura nazionale (National Procedure, NP).
-
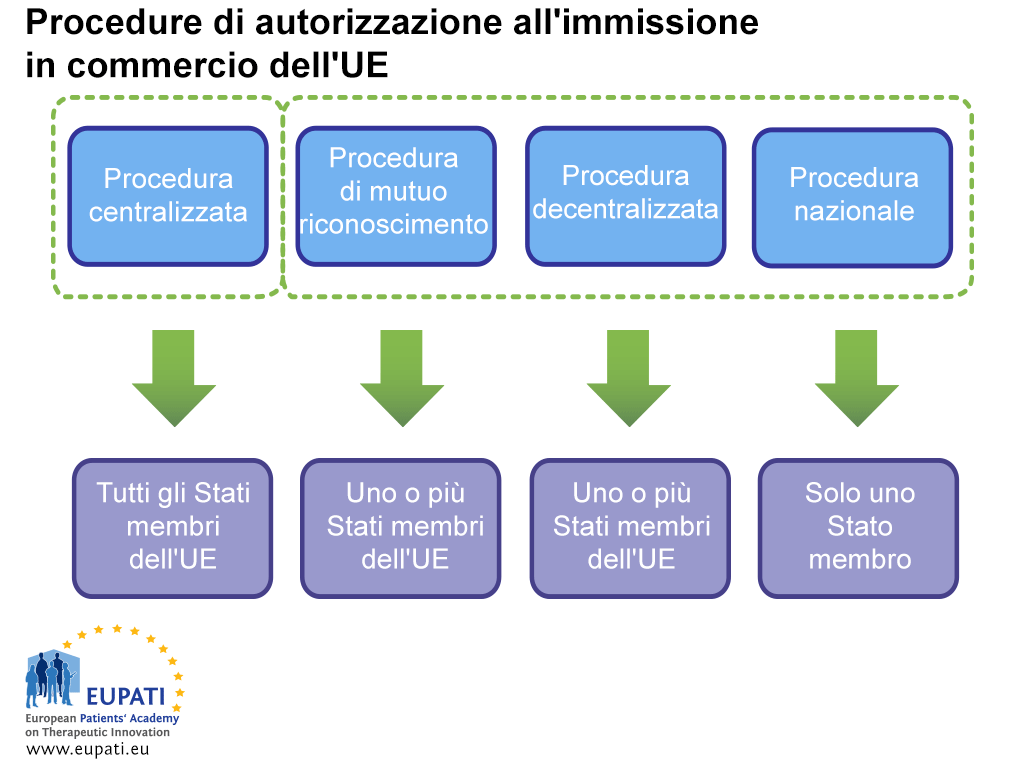
- In funzione della procedura che lo sponsor sceglie o è obbligato a seguire, vi sono diverse parti coinvolte nell’autorizzazione all’immissione in commercio di un farmaco.
Sul sito web dell'EMA possono essere rinvenute risposte a molte delle domande relative alle richieste tramite CP.
Incontri precedenti la presentazione
Gli incontri precedenti la presentazione tra l'azienda e il personale che si occupa di questioni normative si tengono da sei a sette mesi prima della data della presentazione stessa. Tali incontri sono organizzati in modo che l'azienda possa cercare ulteriori informazioni e orientamenti prima di finalizzare il dossier della presentazione.
Per le MAA che seguono la CP, il team del progetto dell'azienda terrà degli incontri con il team dell'EMA che sarà coinvolto nella valutazione della richiesta. Nella MRP, DCP o NP, gli incontri di pre-presentazione con le relative autorità nazionali competenti sono possibili e ugualmente utili.
Presentazione di richiesta di autorizzazione all'immissione in commercio (MAA)
Nella PC, le presentazioni sono possibili soltanto all'EMA in formato eCTD, a meno che non sia concessa un'eccezione. L'eCTD viene concesso tramite un portale online.
Nel caso di richieste seguenti la MRP, DCP o NP, la situazione è più complicata. Tali applicazioni possono coinvolgere fino a 31 agenzie diverse. La rete HMA (Heads of Medicines Agencies network, una collaborazione tra tutti gli Stati membri) offre una soluzione simile all'EMA: la Piattaforma comune europea sulla presentazione (Common European Submission Platform, CESP). Utilizzare il CESP significa che l'azienda deve caricare un dossier sul sistema solo una volta; tutti gli Stati membri coinvolti possono estrarre poi il modulo di presentazione dall'archivio CESP. La piattaforma permette anche di comunicare tra agenzie e il richiedente.
La fase di convalida
Quando l'EMA o l'autorità nazionale competente riceve la presentazione della MAA, il dossier viene prima convalidato per garantire che tutta la documentazione necessaria sia stata inclusa. Quando vi sono domande al richiedente viene data l'opportunità di fornire le risposte necessarie e la documentazione di supporto. Una volta che la MAA è convalidata, inizia la valutazione.
A2-5.11-V1.1