Last update: 5 октября 2016


Введение
Разработка лекарственного препарата — это длительный процесс. Конечной целью любого процесса разработки препарата является получение разрешения на продажу нового лекарственного препарата, регистрационного свидетельства (РС). В фармацевтических компаниях отделы нормативно-правового регулирования (НПР) осуществляют (или должны осуществлять) важную роль на всех этапах жизненного цикла лекарственного препарата. Отделы нормативно-правового регулирования отвечают, в частности, за заявки, которые должны быть поданы в соответствующие органы перед началом проведения каждого клинического исследования, подготовку и подачу досье, которые предоставляются с заявкой на выдачу регистрационного свидетельства (Marketing Authorisation Application, MAA), а также другую деятельность после получения РС, например, за подачу заявок на внесение изменение в регистрационное свидетельство. Специалисты по вопросам нормативно-правового регулирования должны обладать глубокими знаниями в области всех применимых положений по обращению лекарственных препаратов и всего процесса разработки препаратов.
-
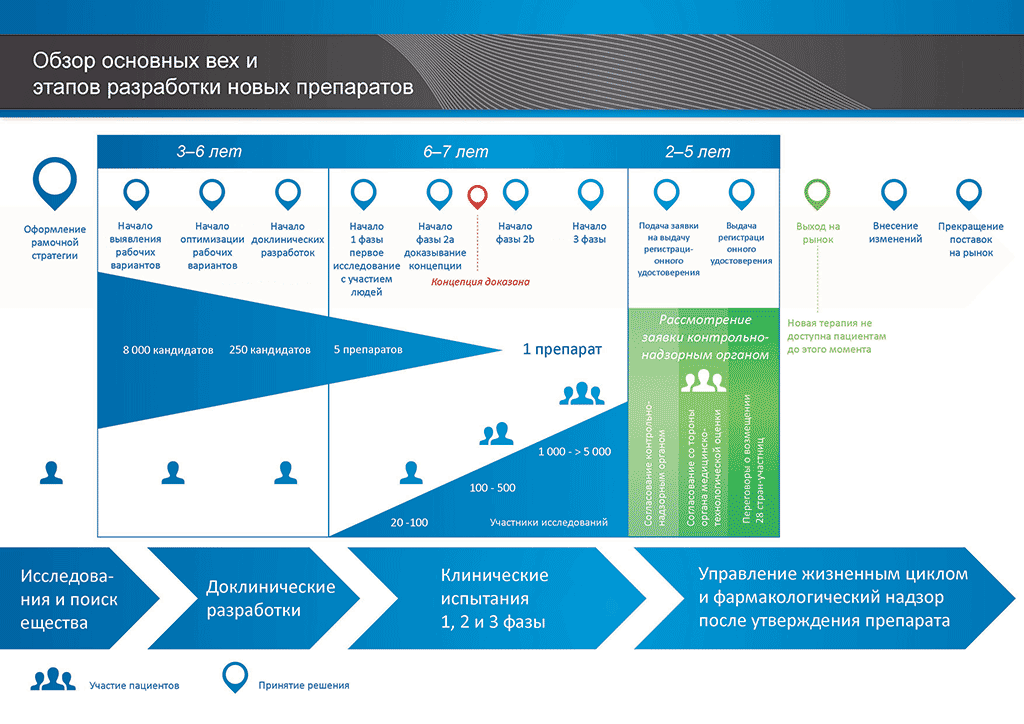
- С момента создания молекулы до момента начала продажи медицинского препарата проходит больше 10 лет, необходимых для тщательнейшего планирования и исследований.
-
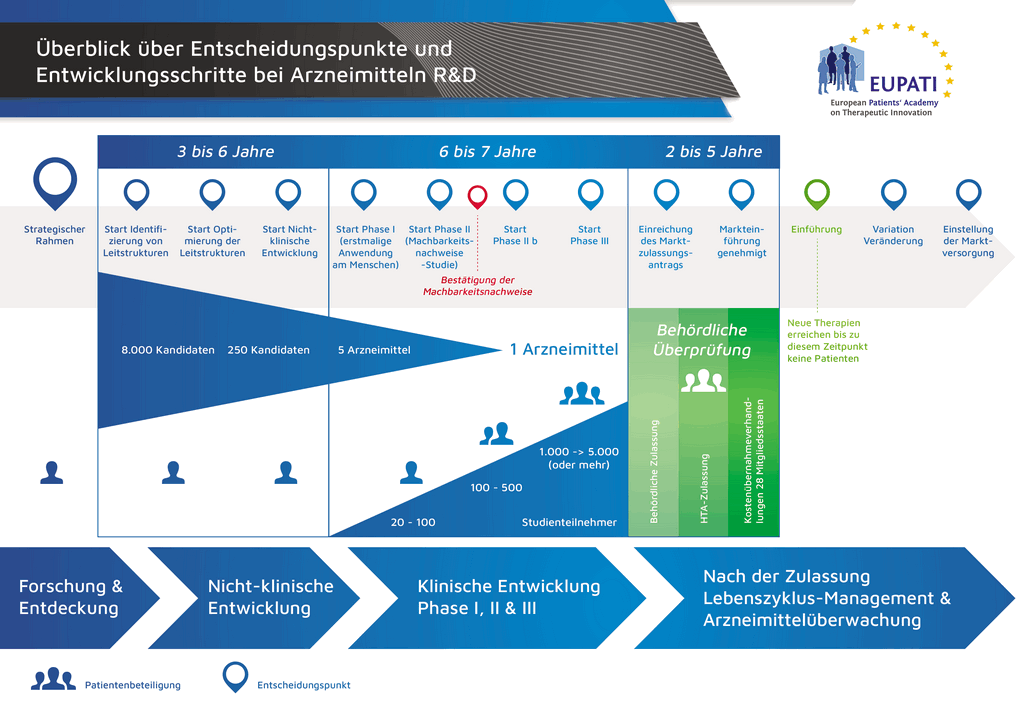
- Es benötigt mehr als 10 Jahre sorgfältiger Planung und Forschung, bis ein Arzneimittel sich vom Molekül zur marktfähigen Behandlung entwickelt hat.
Рисунок 1. Описание процесса разработки лекарственного препарата
Подача заявки на получение регистрационного свидетельства (РС)
На ранней стадии разработки препарата фармацевтическая компания должна принять решение, какой тип заявки на регистрацию препарата ей следует подавать, например:
- полная заявка — смотрите ниже треугольную схему общего технического документа (CTD);
- сокращенная заявка (упрощенная заявка);
- библиографическая заявка: на основании существующей научной литературы.
При подаче заявки требуется предоставление досье документации в соответствующие органы. На рисунке 1 показан процесс разработки нового, инновационного лекарственного препарата. Для этого типа лекарственных препаратов требуется предоставлять полное досье, содержащее все части документации по лекарственному препарату.
Какие части должно включать досье?
На рисунке 2 показаны части общего технического документа (CTD), досье, которое подается в уполномоченные регуляторные органы в виде заявки на выдачу регистрационного свидетельства (МАА) в Канаде, Европе, Японии, Швейцарии, Соединенных Штатах Америки и других странах. Формат CTD был разработан Международным советом по гармонизации технических требований к регистрации лекарственных препаратов для медицинского применения (ICH). CTD также используется для всех типов МАА в ЕС независимо от процедуры (централизованная процедура, ЦП; процедура взаимного признания, ПВП; децентрализованная процедура, ДЦП или национальная процедура, НП) или типа заявки (отдельная, на регистрацию препарата-дженерика и т. д.) Формат CTD применяется для всех типов препаратов (новые химические соединения, радиофармацевтические средства, вакцины, препараты растительного происхождения и т. д.).
Существует пять отдельных модулей CTD.
- Модуль 1: региональная административная информация.
- Модуль 2: резюме и обзоры.
- Модуль 3: качество.
- Модуль 4: отчеты о доклинических исследованиях.
- Модуль 5: отчеты о клинических исследованиях.
модули 2–5 CTD типичны для всех регионов, тогда как модуль 1 специфичен для каждого региона и не считается частью CTD. Большая часть документации по качеству, безопасности и эффективности лекарственного препарата представлена в модулях 3–5. Специалисты по вопросам нормативно-правового регулирования обеспечивают соответствие поданной документации всем релевантным положениям, директивам и рекомендациям. Также они составляют резюме данных, которые включаются в модуль 2.
Рис. 2. Треугольная схема общего технического документа (CTD).
Модуль 1: региональная административная информация
Модуль 1 CTD содержит всю административную информацию, необходимую на региональном уровне. В ЕС существует своя собственная версия модуля 1. Он состоит из следующих 10 элементов:
1.0 Сопроводительное письмо
1.1 Подробное содержание
1.2 Форма заявки
1.3 Информация о препарате
Эта информация будет использоваться как квалифицированными работниками в области здравоохранения, так и пациентами. Она включает в себя краткую характеристику лекарственного средства (КХЛС), подробный документ для квалифицированных работников в области здравоохранения, инструкцию по применению препарата и листок-вкладыш. Должен быть проведен тест на доступность для восприятия, чтобы показать, что листок-вкладыш понятен для лиц, не являющимися специалистами в данной области. Национальные компетентные органы и ЕМА опубликовали на всех языках ЕС образцы информации о препарате, подробно описывающие формат и содержание.
1.4 Информация об экспертах
Модуль 2 CTD содержит резюме и обзоры, составленные экспертами. Каждый из этих экспертов должен предоставить свою профессиональную биографию и подписать заявление о том, что он/она выполнил(-а) правила всех применимых положений или директив при составлении резюме.
1.5 Специфические требования, предъявляемые к разным типам заявки
Дополнительная информация, требуемая для особых типов заявки, таких как библиографические заявки, заявки на регистрацию препарата+-дженерика, «гибридного» препарата или биоаналога, заявка на (расширенный) период исключительного права на данные/продажу, заявка при исключительных обстоятельствах или заявка на выдачу временного регистрационного свидетельства.
1.6 Оценка факторов риска, связанных с окружающей средой
Все действующие вещества, содержащиеся в лекарственных препаратах, могут представлять потенциальный риск для окружающей среды, и все вещества или их метаболиты в конечном счете попадут в окружающую среду. Компания должна рассматривать возможное влияние на окружающую среду в результате использования, хранения и утилизации лекарственного препарата.
1.7 Информация относительно исключительного положения на рынке орфанных лекарственных препаратов
Если лекарственный препарат разработан в качестве орфанного лекарственного препарата для лечения редкого заболевания, требуется особая информация. Если другой препарат на рынке уже занимает исключительное положение по этому же показанию, новый лекарственный препарат может быть одобрен только при особых условиях.
1.8 Информация по фармаконадзору
Должно быть включено описание систем фармаконадзора и управления рисками. Заявитель должен показать функционирование системы надлежащего надзора нежелательных реакций и потенциальных рисков. А также доказательство того, что у заявителя есть квалифицированное лицо, ответственное за фармаконадзор, и также действуют необходимые меры для уведомления о любой нежелательной реакции, возникшей на территории ЕС или другой страны (статья 8 (n) Директивы 2001/83/EC).
1.9 Информация о клинических исследованиях
Заявитель должен включить заявление о том, что клинические исследования лекарственного препарата, проведенные за пределами ЕС, отвечают требованиям ЕС.
1.10 Информация о педиатрии
Все новые лекарственные препараты в ЕС должны рассматриваться для применения в популяции детей. Как правило, они должны пройти исследования у детей. Однако по этому требованию возможен отказ, если заболевание отмечается только у пожилых и взрослых больных, или если новый лекарственный препарат, вероятно, неэффективен или небезопасен в некоторой части или во всей популяции детей. Если отказа нет, компания должна составить план исследования у детей (Paediatric Investigation Plan, PIP) за исключением случаев, когда выдана отсрочка, тогда этот план может быть составлен позже. Данный раздел должен включать копию отказа или решения по плану исследования у детей (включая отсрочки в применимых случаях).
Как собирается досье?
В большинстве случаев уже невозможно подавать заявки на регистрацию в бумажной форме, это значит, что вся документация по пяти модулям CTD должна подаваться в типовом электронном формате: eCTD. Электронный CTD представляет собой не просто пакет документов или один большой файл в формате PDF. Скорее электронный CTD — это стандарт, в котором подробно описывается пакет и структура файлов, которые должны быть соблюдены для того, чтобы компания и регуляторные органы могли легко ориентироваться в досье ровно как и Вы в обычном компьютерном каталоге.
Что представляет собой процесс подачи пакета документов на регистрацию препарата?
Заявитель должен подробно рассмотреть все логистические и регуляторные вопросы перед подачей заявки. Сюда относится выбор процедуры подачи заявки на выдачу регистрационного свидетельства: децентрализованная процедура (ДЦП), процедура взаимного признания (ПВП), децентрализованная процедура (ДЦП) и национальная процедура (НП).
-
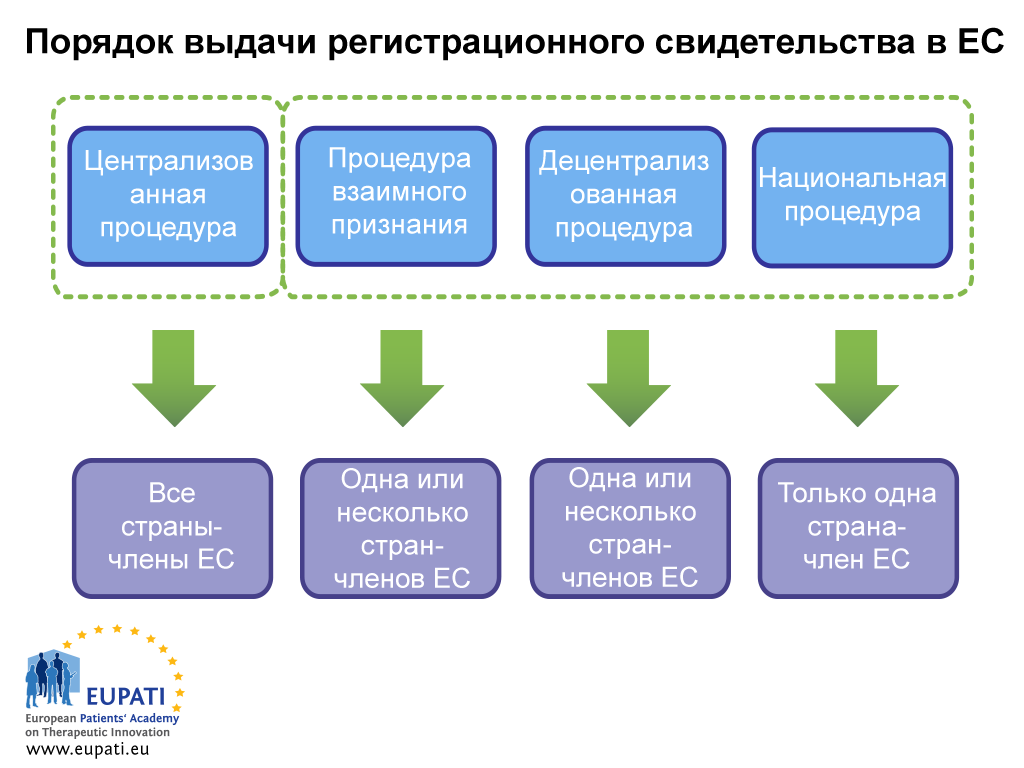
- зависимости от того, какую процедуру выбирает спонсор (или какой он вынужден следовать), процесс получения регистрационного свидетельства медицинского препарата может требовать участия разных сторон.
Ответы на многие вопросы относительно подачи заявки в соответствии с ЦП можно найти на веб-сайте ЕМА.
Встречи до подачи документов
Встречи до подачи документов на регистрацию между компанией и персоналом регуляторных органов происходят за шесть-семь месяцев до даты подачи документов. Эти встречи организовываются таким образом, чтобы компания могла найти дополнительную информацию и руководства перед завершением досье на регистрацию препарата.
По заявкам на регистрацию препарата в соответствии с ЦП группа проекта от компании должна встретиться с группой от ЕМА, которая будет задействована в процессе оценки заявки. При ПВП, ДЦП или НП встречи до подачи документов с соответствующими национальными компетентными органами возможны и в равной степени полезны.
Подача заявки на выдачу регистрационного свидетельства (МАА)
При ЦП подача заявки возможна в ЕМА только в формате электронного CTD, кроме случаев исключений. Электронный CTD подается через онлайн портал.
В случае подачи заявки в соответствии с ПВП, ДЦП или НП ситуация более сложная. В процесс подачи таких заявок может быть задействовано до 31 разного агентства. В настоящее время сеть HMA (глав агентств по лекарственным средствам, сотрудничество между государствами-членами ЕС) предлагает ЕМА аналогичное решение: Общую европейскую платформу для подачи заявок на регистрацию (Common European Submission Platform, CESP). Использование CESP означает, что компании необходимо всего лишь сразу загрузить досье в систему, затем все государства-члены ЕС, задействованные в данном процессе, могут извлечь форму подачи заявки из базы данных CESP. Эта платформа позволяет также агентствам и заявителю обмениваться информацией.
Фаза валидации
При получении ЕМА или национальным компетентным органом пакета документов на регистрацию препарата сначала валидируется досье для проверки того, что досье содержит всю необходимую документацию. При возникновении вопросов заявителю дается возможность предоставить необходимые ответы и пояснительную документацию. После валидации МАА начинается проведение оценки.
A2-5.11-V1.1