

Introduction
Le processus de développement des médicaments est une aventure au long cours. La finalité ultime de tout processus de développement est l’obtention d’une autorisation de commercialisation du nouveau produit pharmaceutique : une autorisation de mise sur le marché (AMM). Au sein des laboratoires pharmaceutiques, le service des affaires réglementaires constitue (ou devrait constituer) une partie intégrante de toutes les étapes du cycle de vie du produit pharmaceutique. Le service des affaires réglementaires est notamment responsable des demandes qui doivent être déposées avant tout essai clinique, de la préparation et la soumission du dossier de demande d’autorisation de mise sur le marché (DAMM) et d’autres activités après octroi de l’AMM, telles que les demandes de modification de l’AMM (variation). Les professionnels des affaires réglementaires doivent posséder une connaissance approfondie de toutes les réglementations applicables dans le domaine du médicament, ainsi que de l’ensemble du processus de développement.
-

- Il faut compter plus de 10 ans de planification et de recherche minutieuses pour qu’un médicament passe de l’état de molécule à celui de traitement commercialisable.
Figure 1 : présentation générale du processus de développement des médicaments
Dossiers de demande d'autorisation de mise sur le marché (AMM)
Le laboratoire pharmaceutique doit décider à un stade précoce du développement du type de demande à présenter pour l'autorisation de mise sur le marché, par exemple :
- Dossier de demande complet (voir le triangle du document technique commun (CTD) ci-dessous)
- Dossier de demande abrégé (demande réduite)
- Demande bibliographique, basée sur la documentation scientifique existante
Les demandes nécessitent le dépôt d'un dossier de documentation auprès des autorités concernées. La figure 1 illustre le processus de développement d'un nouveau médicament innovant. Ce type de médicament requiert la soumission d'un dossier complet, dans lequel tous les éléments de documentation du médicament doivent être inclus.
Quels sont les éléments à inclure dans un dossier ?
La figure 2 représente les éléments qui composent le document technique commun (CTD), dossier déposé auprès des autorités réglementaires comme demande d'autorisation de mise sur le marché (DAMM) au Canada, en Europe, au Japon, en Suisse, aux États-Unis et dans d'autres pays. Le format du CTD a été mis au point par le Conseil international d'harmonisation des exigences techniques pour l'enregistrement des médicaments à usage humain (ICH). Le CTD doit être utilisé pour tous les types de demandes d'AMM au sein de l'UE indépendamment de la procédure (procédure centralisée PC, procédure de reconnaissance mutuelle PRM, procédure décentralisée PDC ou procédure nationale PN) ou du type de demande (autonome, générique, etc.). Le format du CTD s'applique à tous les types de produits (nouvelles entités chimiques, produits radiopharmaceutiques, produits à base d'herbes, etc.).
Le format du CTD se compose de cinq modules distincts.
- Module 1 : Informations administratives régionales
- Module 2 : Résumés et présentations générales
- Module 3 : Qualité
- Module 4 : Rapports d'études non cliniques
- Module 5 : Rapports d'études cliniques.
Les modules 2 à 5 du CTD sont communs à toutes les régions, tandis que le module 1 est spécifique à chaque région et n'est pas considéré comme faisant partie du CTD. La majeure partie de la documentation relative à la qualité, la sécurité et l'efficacité du médicament est contenue dans les modules 3 à 5. Les professionnels des affaires réglementaires garantissent que la documentation soumise est conforme à toutes les réglementations, directives et lignes directrices applicables. Ils produisent également les résumés inclus dans le module 2.
Figure 2 : triangle du document technique commun.
Module 1 : Informations administratives régionales
Le module 1 du CTD contient toutes les informations administratives nécessaires à un niveau régional. L'UE possède sa propre version du module 1, qui se compose des 10 éléments suivants :
1.0 Lettre de présentation
1.1 Sommaire complet
1.2 Formulaire de demande
1.3 Informations sur le produit
Il s'agit des informations qui seront utilisées à la fois par les professionnels de santé et les patients. Elles incluent le Résumé des caractéristiques du produit (RCP), un document détaillé destiné aux professionnels de santé, l'étiquetage et la notice. Un test de lisibilité doit être effectué pour démontrer que la notice peut être comprise par des personnes non initiées. Les autorités nationales compétentes et l'EMA ont publié des modèles dans toutes les langues de l'UE pour la présentation des informations sur les produits qui fournissent des indications détaillées sur le format et le contenu prescrits.
1.4 Informations sur les experts
Le module 2 du CTD contient des résumés et des présentations rédigés par des experts. Chacun de ces experts doit fournir un curriculum vitae (CV) et signer une déclaration établissant qu'il a respecté les règles de toutes les réglementations ou directives applicables dans la rédaction des résumés.
1.5 Exigences spécifiques pour les différents types de demandes
Informations supplémentaires requises pour des types spéciaux de demande, telles que les demandes bibliographiques, génériques, « hybrides » ou biosimilaires, d'exclusivité de données/commercialisation (étendue), la demande dans des circonstances exceptionnelles ou la demande d'autorisation de mise sur le marché conditionnelle.
1.6 Évaluation des risques environnementaux
Toutes les substances actives contenues dans les médicaments peuvent présenter un risque pour l'environnement, et toutes les substances ou leurs métabolites aboutissent finalement dans l'environnement. La société doit traiter le problème de l'impact environnemental possible créé par l'utilisation, le stockage et l'élimination du médicament.
1.7 Informations relatives à l'exclusivité commerciale du médicament orphelin
Des informations spéciales sont requises si le médicament est désigné comme médicament orphelin destiné à traiter une maladie rare. Si un autre médicament sur le marché possède déjà l'exclusivité commerciale pour la même indication, le nouveau médicament peut être approuvé uniquement dans des conditions spéciales.
1.8 Informations relatives à la pharmacovigilance
Une description des systèmes de pharmacovigilance et de gestion des risques doit être incluse. Le demandeur doit démontrer qu'une surveillance appropriée des réactions indésirables et des risques potentiels est en place. Cela doit inclure la preuve que le demandeur dispose d'une personne qualifiée chargée de la pharmacovigilance et des moyens nécessaires pour la notification de toute réaction indésirable survenant soit dans l'Union européenne, soit dans un pays tiers (Article 8 (n) de la Directive 2001/83/CE).
1.9 Informations relatives aux essais cliniques
Le demandeur doit inclure une déclaration stipulant que tous les essais cliniques du médicament menés en dehors de l'UE sont conformes aux exigences de l'UE.
1.10 Informations relatives à la pédiatrie
Dans l'UE, tous les nouveaux médicaments doivent être envisagés pour la population pédiatrique. En général, ils doivent être testés chez les enfants. Toutefois, une dérogation à cette obligation peut être accordée si la maladie est observée uniquement chez les personnes âgées ou les adultes, ou s'il est vraisemblable que le nouveau médicament est inefficace ou dangereux pour une partie ou l'ensemble de la population pédiatrique. En l'absence de dérogation, la société doit produire un plan d'investigation pédiatrique (PIP), sauf si un report a été accordé, auquel cas le PIP peut être produit ultérieurement. Cette section doit inclure une copie de la dérogation ou de la décision relative au PIP (y compris les reports, le cas échéant).
Comment le dossier est-il constitué ?
Dans la plupart des cas, les soumissions sous forme papier ne sont plus possibles. Cela signifie que toute la documentation associée aux cinq modules de CTD doit être soumise dans un format électronique normalisé : l'eCTD. L'eCTD n'est pas juste une collection de documents PDF, ni un volumineux fichier PDF unique. L'eCTD est une norme qui décrit en détail la structure des dossiers et des fichiers qu’il convient de respecter pour permettre à la société et aux autorités réglementaires de naviguer facilement dans le dossier, comme dans un répertoire d'ordinateur normal.
En quoi consiste le processus de soumission ?
Le demandeur doit examiner soigneusement tous les aspects logistiques et réglementaires avant la soumission. Cela inclut le choix de la procédure de demande d'autorisation de mise sur le marché à utiliser : la procédure centralisée (PC), la procédure de reconnaissance mutuelle (PRM), la procédure décentralisée (PDC) ou la procédure nationale (PN).
-
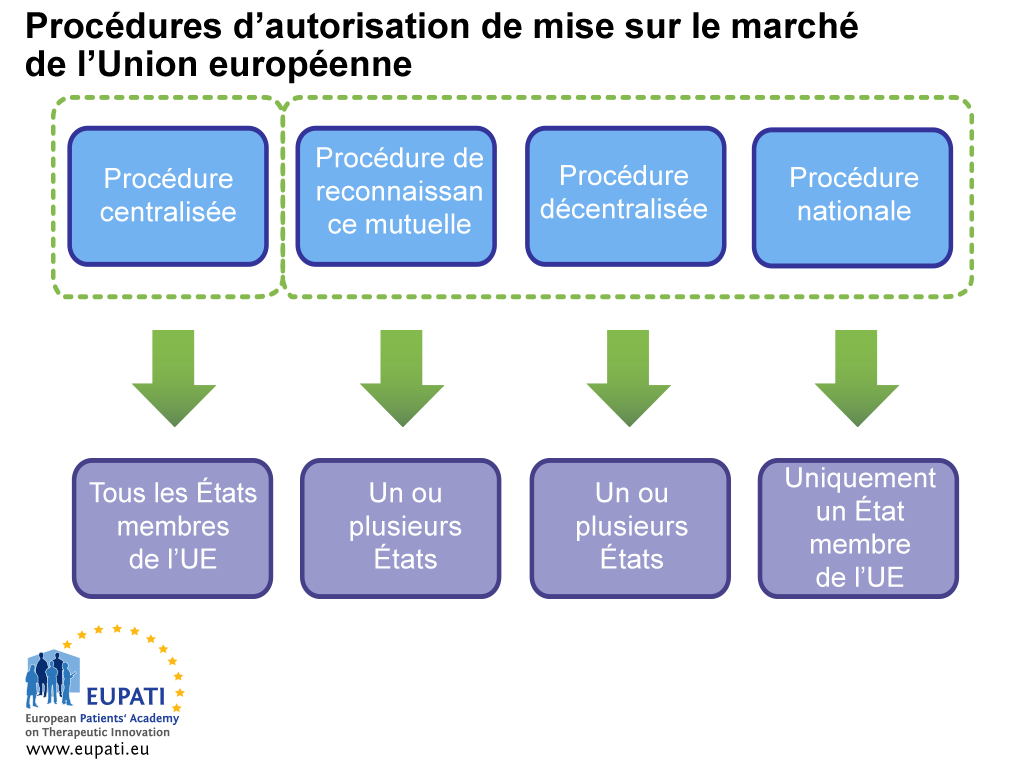
- Différents acteurs sont impliqués dans l’autorisation de mise sur le marché d’un médicament selon la procédure choisie par le promoteur ou celle qu’il est obligé de suivre.
Des réponses à de nombreuses questions relatives aux demandes par le biais de la procédure PC sont consultables sur le site Web de l'EMA.
Réunions préalables à la soumission
Des réunions préalables à la soumission entre la société et le personnel réglementaire ont généralement lieu six à sept mois avant la date de la soumission. Ces réunions sont organisées pour permettre à la société d'obtenir des informations et des orientations supplémentaires avant de finaliser le dossier de demande.
Pour les autorisations d'AMM via la procédure centralisée (PC), l'équipe de projet de la société se réunit avec l'équipe de l'EMA qui participera à l'évaluation de la demande. Dans les procédures PRM, PDC ou PN, les réunions préalables à la soumission avec les autorités nationales compétentes concernées sont possibles et également utiles.
Soumission de la demande d'autorisation de mise sur le marché (DAMM)
Dans la procédure centralisée, les demandes doivent être déposées auprès de l'EMA uniquement dans le format eCTD, sauf si une exception a été accordée. Le dossier eCTD est soumis par l'intermédiaire d'un portail en ligne.
Dans le cas des demandes selon les procédures PRM, PDC ou PN, la situation est plus compliquée. Ces demandes peuvent impliquer jusqu'à 31 agences différentes. Le réseau HMA (réseau des chefs d'agences des médicaments, collaboration entre tous les États membres) offre à présent une solution similaire à celle de l'EMA : la plateforme de soumission européenne centralisée (CESP). Lorsque la voie de la CESP est utilisée, il suffit pour la société d'importer une seule fois un dossier dans le système et tous les États membres peuvent ensuite extraire la soumission à partir du référentiel CESP. Cette plateforme permet également la communication entre les agences et le demandeur.
La phase de validation
Lorsque l'EMA ou l'autorité nationale compétente reçoit la soumission de demande d'AMM, le dossier est d'abord validé pour vérifier qu'elle comporte toute la documentation nécessaire. En cas de questions, le demandeur a la possibilité de fournir les réponses nécessaires et les pièces justificatives. Une fois la demande d'AMM validée, l'évaluation commence.
A2-5.11-V1.1