

Einleitung
Der nicht-klinische (oder vorklinische) Entwicklungsabschnitt befasst sich primär mit der Identifizierung derjenigen potenziellen Therapie, die den größten Erfolg verspricht, der Beurteilung der Sicherheit dieser Therapie, und dem Aufbau umfassender und belastbarer wissenschaftlicher Erkenntnisse vor dem Übergang in den klinischen Entwicklungsabschnitt.
Des Weiteren fallen gewisse nicht medizinische Aktivitäten in den nicht-klinischen Entwicklungsabschnitt, beispielsweise die Festlegung, ob und in welchem Umfang für den Wirkstoffkandidaten Ansprüche auf geistiges Eigentum angemeldet werden, aber auch die Herstellung ausreichender Mengen des Wirkstoffs für klinische Studien. Der nicht-klinische Entwicklungsabschnitt eines Arzneimittels ist komplex und unterliegt zulassungsrechtlichen Vorgaben.
Grundlagen, wesentliche Definitionen und Konzepte
„Nicht-klinisch“ oder „vor-klinisch“?
Die Begriffe „nicht-klinisch“ und „präklinisch“ werden oftmals synonym verwendet.
Wiewohl diese in den präklinischen Phasen der Entwicklung unverzichtbar sind, können nicht-klinische Studien jederzeit während des Lebenszyklus des Produkts durchgeführt werden. Es empfiehlt sich jedoch, dies so früh wie möglich zu tun, um Überraschungen in späteren Phasen der Entwicklung zu vermeiden.
Über die Identifizierung von Pharmakodynamik (Was das Arzneimittel mit dem Körper macht), Pharmakokinetik (Was der Körper mit dem Arzneimittel macht) und Toxikologie des Wirkstoffkandidaten vor der Anwendung am Menschen hinaus dienen die Daten aus nicht-klinischen Studien zur Verfeinerung, Konsolidierung und Ergänzung von Informationen zum Zweck der Aktualisierung des Sicherheitsprofils des Produkts in der präklinischen Phase, zum Zeitpunkt der Zulassung und während des Lebenszyklus des Arzneimittels.
in-silico-, in-vitro– und in-vivo-Studien
Im Rahmen der nicht-klinischen Entwicklung werden Studien der folgenden Art durchgeführt:
- in-silico: Auf einem Computer oder mittels einer Computersimulation durchgeführte Studie, beispielsweise zur Vorhersage des toxikologischen Profils eines Produkts unter Verwendung datenbankbasierter Ansätze und auf Grundlage seiner chemischen Struktur.
- in-vitro (lateinisch: „Im Glas“): Durchführung von Versuchen in einer kontrollierten Umgebung außerhalb eines lebenden Organismus, beispielsweise die Verwendung von Hepatozytenkulturen (Leberzellen) für Untersuchungen zur Verstoffwechselung.
- in-vivo (lateinisch: „Im Lebenden“): Experimente an einem vollständigen, lebenden Organismus (z. B. Tiere, Menschen oder Pflanzen) statt nur an isolierten Geweben oder Zellen
Welche Schlüsselaspekte umfasst der Abschnitt „Chemie, Herstellung und Qualitätskontrolle“ (Chemistry, Manufacturing, Control, CMC) in der nicht-klinischen Entwicklung?
Alle Studien im Rahmen der nicht-klinischen Entwicklung sind auf die Produktion eines geeigneten Wirkstoffs angewiesen:
- Für nicht-klinische Studien werden nur geringe Mengen (Milligramm- bis Gramm-Bereich) benötigt; im Anschluss gilt es dann, einen Scale-Up-Prozess zu entwickeln, um größere Mengen für klinische Studien und später – nach der Zulassung – für den Markt zu produzieren.
- Für GLP-Studien (Good Laboratory Practice, Gute Laborpraxis) werden qualifizierte oder GMP-Chargen (Good Manufacturing Practice, Gute Herstellungspraxis) des Wirkstoffs benötigt.
Zu den Schlüssel-CMC-Schritten der nicht-klinischen Entwicklungsphase zählen u. a.:
- Bestimmung der Dosis und des Verabreichungswegs
- Detaillierte physiko-chemikalische Charakterisierung
- Stabilitätsprüfungen und Verunreinigungsanalysen
- Entwicklung und Validierung von Methoden für die Quantifizierung des Wirkstoffs in Körperflüssigkeiten wie Blut, Plasma und Harn in Studien zu Aktivität und Nebenwirkungen
- Entwicklung eines Prototypen für das in der Klinik Verwendete
Der nicht-klinische Entwicklungsprozess
Die Aktivitäten der nicht-klinischen Entwicklung erfolgen parallel zu den Forschungsaktivitäten. Sie sind im Folgenden skizziert und sollen das geplante klinische Entwicklungsprogramm unterstützen.
Zielsetzungen
Nach Identifizierung eines Wirkstoffkandidaten muss die nicht-klinische Entwicklung damit beginnen, die folgenden Fragen zu beantworten, wobei die Antworten aus spezifischen Beurteilungen/Studien resultieren:
- Wirkt er? → Wirksamkeitsbeurteilung
- Wie wird er verabreicht, und wie wird der Körper reagieren? → Profilerstellung
- Ist er sicher? → Toxikologie/Sicherheit
- Ist die Produktion realisierbar und steuerbar?
Nicht-klinische Entwicklungsaktivitäten können sich über den gesamten Lebenszyklus des Produkts hinweg fortsetzen, wobei jedoch gilt: je früher diese Fragen beantwortet werden, desto leichter ist es, das Profil des Patienten zu identifizieren, der den größten Nutzen ziehen wird.
Projekt-Management
Das nicht-klinische Entwicklungsprogramm ist komplex und erfordert ein verlässliches Projekt-Management und Kommunikationsfertigkeiten bei der Anleitung multidisziplinärer Teams. Das Projektteam muss den vorgesehenen klinischen Plan verstehen, um den nicht-klinischen Plan und die zugehörigen Aktivitäten festlegen zu können.
Das Profil bietet ein Gerüst für die Durchführung der nicht-klinischen Entwicklungsstrategie, die Festlegung von Zielen, Risiken, Verantwortlichkeiten und die Go/No-Go-Entscheidungsfindung. Die Profil-Implementierung trägt dazu bei, den Fokus des Projekts auf relevante Produktkriterien gerichtet zu halten, um Go/No-Go-Entscheidungen zeitnah fällen und das Gesamtprojektrisiko (d. h. die Weiterführung der Entwicklung eines nicht zweckmäßigen Produkts) zu reduzieren.
Nicht-klinische gesetzliche Leitlinien
Viele Akteure sind an der Entwicklung von Arzneimitteln beteiligt, und jede Organisation oder Institution folgt ihren eigenen Regeln. Für Unternehmen beispielsweise sind dies ihre standardisierten Abläufe (SOPs, Standard Operating Procedures). Ergänzend zu den Vorkehrungen der „Guten klinischen Praxis“ (GCP) finden sich auf der Website der Europäischen Arzneimittel-Agentur (EMA) weitere Leitlinien.
- Hierbei handelt es sich entweder um allgemeine oder um spezifische wissenschaftliche und technische Aspekte (spezifisch beispielsweise hinsichtlich der erforderlichen toxikologischen Studien).
- Bei jedem neuen Antrag auf Marktzulassung sind diese genauestens einzuhalten; jede Abweichung muss begründet werden.
Die Darstellung der Daten erfolgt im CTD-Format (Common Technical Document, Standardisierter Antrag auf Zulassung eines Arzneimittels), das von der ICH (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Internationale Konferenz zur Harmonisierung der Beurteilungskriterien von Human-Arzneimitteln als Basis der Arzneimittelzulassung) definiert worden ist. Die Vereinbarung, sämtliche Informationen zu Qualität, Sicherheit und Wirksamkeit in diesem standardisierten Format (CTD) zusammenzustellen, hat den Prozess der behördlichen Überprüfung (Zulassung) revolutioniert und zu harmonisierten elektronischen Einreichungen geführt, die wiederum die Implementierung Guter Begutachtungs-Praktiken (Good Review Practices) ermöglicht haben. Für die Industrie hat diese Harmonisierung die Notwendigkeit beseitigt, die Informationen für die Einreichung bei den verschiedenen Zulassungsbehörden immer wieder neu zu formatieren (die ICH hat die Zulassungsbehörden und pharmazeutischen Unternehmen Europas, Japans und der USA an einen Tisch gebracht, um hinsichtlich der wissenschaftlichen und technischen Aspekte der Zulassung von Arzneimitteln ein einheitliches Vorgehen zu vereinbaren).
-
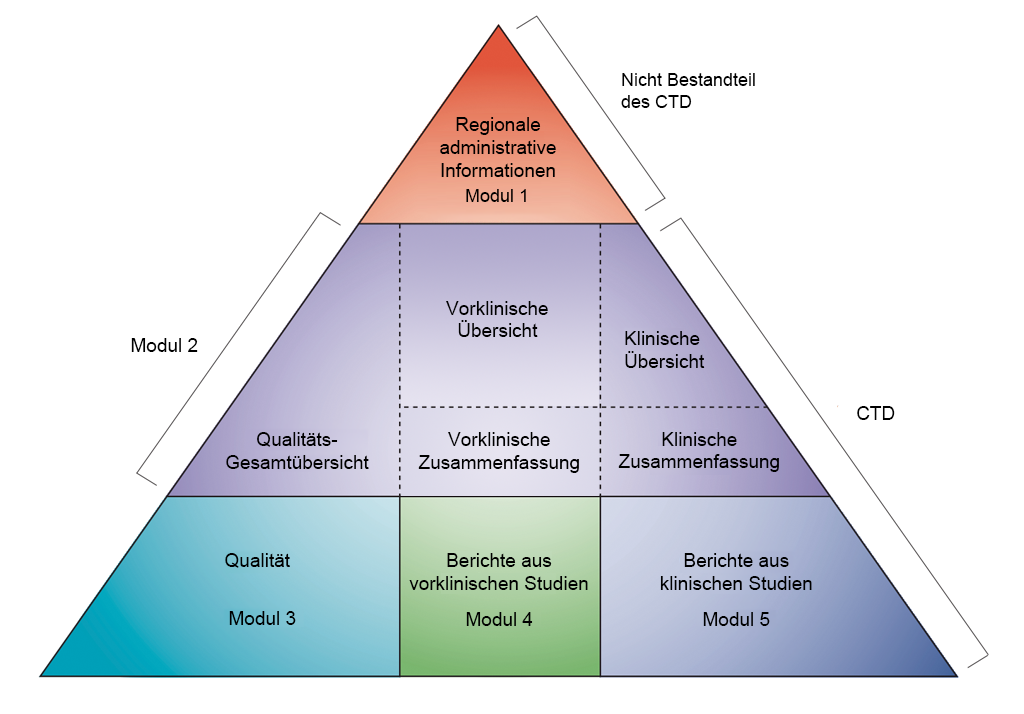
- Nicht-klinische Entwicklung in CTD-Modulen. Übernommen von ICH CTD (siehe Referenz 1)
Das CTD ist in fünf Module unterteilt (siehe vorstehende Abbildung). Im Juli 2003 wurde das CTD das obligatorische Format für neue Anträge auf Marktzulassung in der EU und in Japan sowie das bevorzugte Format für die Einreichung von NDA-Anträgen (New Drug Applications) bei der US-amerikanischen Gesundheitsbehörde FDA (Food and Drug Administration).
Zusammenfassung
Die nicht-klinische Entwicklungsphase ist von großer Bedeutung; sie muss potentiellen Problemen zuvorkommen, bevor ein Wirkstoff in die klinische Entwicklung überführt wird.
Für die Durchführung von klinischen Studien mit einem Wirkstoffkandidaten müssen verschiedene Voraussetzungen erfüllt sein:
- Erfolgte Durchführung der nicht-klinischen Sicherheitsbeurteilung unter GLP-Bedingungen (Gute Laborpraxis)
- Durchführung der Herstellung unter angemessener Qualitätskontrolle
- Dokumentierung der Daten und des Prozesses im CTD-Format und Erstellung der Grundlagen für die klinische Entwicklungsphase
Es ist eine zunehmende Tendenz zu beobachten, arzneimittelähnliche Eigenschaften in silico zu entwerfen. Auch die Anwendung von Methoden der Bioinformatik für die Modellierung und Vorhersage erfreut sich zunehmender Beliebtheit.
Die Richtlinien für die nicht-klinische Entwicklung sind Gegenstand kontinuierlicher Harmonisierungsbestrebungen (unter besonderer Berücksichtigung von Sicherheit und Qualität) der wichtigsten Zulassungsbehörden (Europa, USA und Japan): Die ICH gibt regelmäßig detaillierte Leitfäden (vergleichbar denen, die von der Europäischen [EMA] und der US-amerikanischen Zulassungsbehörde [FDA] herausgegeben werden) für die pharmazeutische Industrie heraus.
Weitergehende Informationen
- European Medicines Agency (2007a). Guideline on strategies to identify and mitigate risks for first-in human trials with investigational medicinal products. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf
- European Medicines Agency (2007b). Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002989.pdf
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
- European Medicines Agency (2011). Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
Quellenangaben
- Image reproduced from ICH (2015). M4: The Common Technical Document. Retrieved 11 July, 2021, from https://www.ich.org/page/ctd
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf