Last update: 15 7月 2015


はじめに
非臨床 (前臨床) 開発段階の主な目的は、臨床開発段階に移行する前に、候補となる治療法の中で最も成功する確率が高いものを特定し、その安全性を評価し、確固たる科学的根拠を確立することです。
また非臨床開発段階では、候補化合物の非医学的な目的を満たす必要があります。たとえば、知的財産権を定義したり、治験で使用する十分な量の医薬品を製造したりします。医薬品の非臨床開発は複雑であり、規制によって左右されます。
基本、主な定義、概念
「非臨床」か「前臨床」か?
「非臨床」と「前臨床」は、同義で使われることが多い用語です。
開発の前臨床段階には重大な関連性がありますが、非臨床試験は製品のライフサイクルのどの時点でも実施できます。ただし、開発後期に不測の事態が発生するのを防ぐため、ほとんどの試験はできるだけ早期に実施することが望まれます。
薬力学 (医薬品が身体に及ぼす影響)、薬物動態 (医薬品に対する身体の反応)、およびヒトに投与する前の候補化合物の毒物学の同定を越えて、非臨床試験から得られたデータは洗練され、まとめられるために使用され、前臨床段階の間や登録時、医薬品のライフサイクル中に製品の安全性プロフィールを更新するための情報を追加します。
コンピューター上、試験管内、生体内試験
非臨床開発における試験は、次の方法で実施します。
- In silico (コンピューター上):「コンピューター上またはコンピューター シミュレーションを通じて実施」します。たとえば、データをベースとしたアプローチから得られた製品の化学構造を使用して、その製品の毒性プロフィールを予測します。
- In vitro (試験管内) (ラテン語で「ガラスの中」という意味):生体外の管理環境下で試験手順を実行します。たとえば、肝細胞を培養して代謝の試験を行います。
- In vivo (生体内) (ラテン語で「生物の中」という意味):組織や細胞ではなく、完全な生体 (動物、ヒト、植物) を使用して実験を行います。
非臨床開発における化学、製造および品質管理 (CMC) の主要な側面とは何ですか?
すべての非臨床開発試験では、適切な量の活性物質を製造する必要があります。
- 非臨床試験で必要とされる量は、通常は少量 (数ミリグラムから数グラム) です。その後の治験や承認後の市販に向け、大量に生産するため規模拡大プロセスを開発する必要があります。
- 医薬品の安全性試験の実施基準 (GLP) 試験では、適切な量、つまり医薬品の製造管理および品質管理の基準 (GMP) に従った量の活性物質が必要になります。
非臨床開発段階における主要な CMC 手順の例としては、以下のものが挙げられます。
- 用量および投与方法の特定
- 物理化学特性の詳細
- 安定性試験および不純物分析
- 活性試験および副作用試験において、血液、血漿、尿などの体液に含まれる活性物質を定量化する手法の開発および検証
- 施設で使用するプロトタイプの開発。
非臨床開発のプロセス
非臨床開発の活動は研究活動に類似しています。以下に示す目的と質問に取り組み、計画されている臨床開発プログラムを支持しなければなりません。
目的
候補化合物を特定したら、非臨床開発によって以下の質問に答えられるようにします。答えは、具体的な評価や試験から導き出します。
- 効き目はありますか?→ 有効性評価
- 医薬品が体内でどのように送達され、身体がどのように反応しますか?→ プロファイリング
- 安全ですか?→ 毒性学/安全性
- 製造は実行可能で管理可能ですか?
非臨床開発の活動は製品のライフサイクル全体を通じて継続できますが、質問に答える時期が早いほど、最も大きなベネフィットを得られる患者のプロフィールを特定しやすくなります。
プロジェクト管理
非臨床開発プログラムは複雑であり、多数の領域にわたるチームを活用するには、確固たるプロジェクト管理スキルとコミュニケーション スキルが要求されます。プロジェクトチームは、非臨床計画と関連する活動を定義するために、意図された臨床計画について理解しておく必要があります。
非臨床開発戦略を実施するフレームワークとなるのがプロフィールであり、目標やリスク、責任、測定指標、実行か中止かを決定する基準を定義します。プロフィールを取り入れることで、プロジェクトの焦点を主要な製品基準に置くことができるほか、「実行か中止か」の意思決定をタイムリーに実行したり、全体的なプロジェクトのリスク (有用ではない製品の開発継続) を低減したりできます。
非臨床開発の規制ガイドライン
医薬品の開発には多くの人が関わり、各組織や機関はそれぞれで定めた規則に従って行動します。たとえば、各企業は独自の標準業務手順 (SOP) を定めています。医薬品の臨床試験の実施に関する基準の規定に加え、欧州医薬品庁 (EMA) のウェブサイトではガイドラインを参照することができます。
- このような規定は、科学的および技術的な側面に対処した全般的または具体的な内容です (例: 必要とされる毒性試験に特有のもの)。
- このような規定は、新しい販売承認申請に厳密に従ったものでなければならず、逸脱した部分については正当な理由を明示する必要があります。
データの形式は、医薬品規制調和国際会議 (ICH) で定義されたコモン テクニカル ドキュメント (CTD) 形式に従うものとします。品質、安全性、有効性に関する情報をすべて共通の形式 (CTD) でまとめるという合意により、承認審査のプロセスに大きな変化がもたらされ、統一された形式での電子申請につながり、優れた審査の実施を可能にしています。業界にとっては、提出する情報の形式を規制当局ごとに再構成する必要性がなくなりました (ICH では、欧州、日本、米国の規制当局と製薬業界各社が集まり、医薬品登録の科学的および技術的側面について議論しています)。
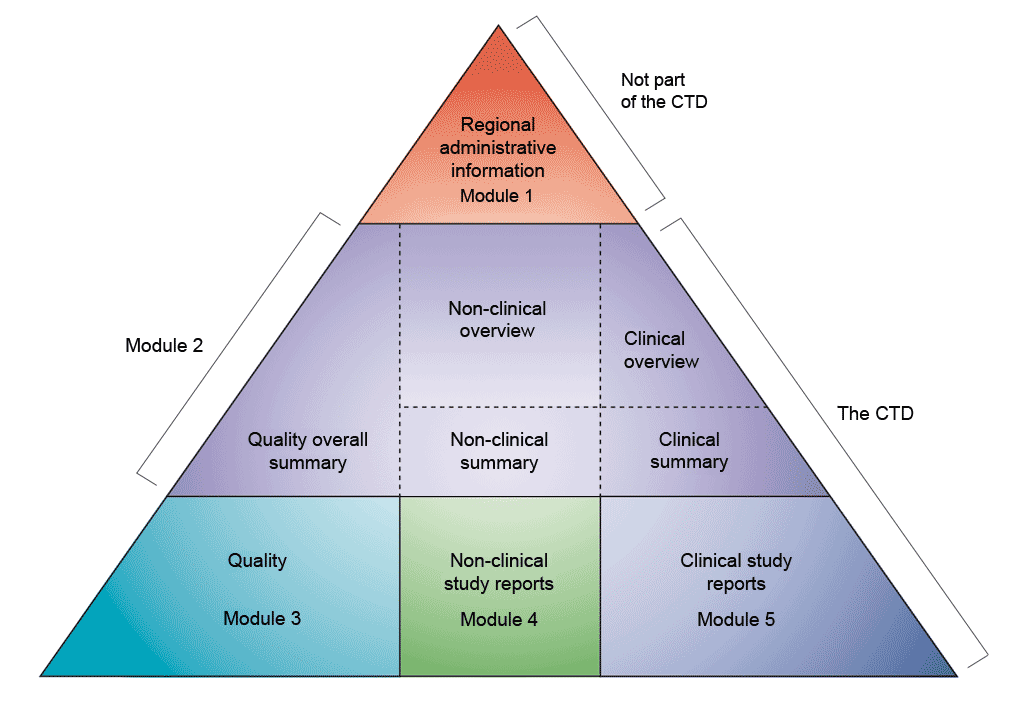
CTD は、5 つのモジュールに編成されています (上記の図を参照)。2003 年 7 月、欧州および日本における新規販売承認申請については、CTD が必須の形式となりました。また、米国食品医薬品局 (FDA) に提出する新薬承認申請 (NDA) の形式としても強く勧告されています。
概要
非臨床開発段階は非常に重要であり、化合物を臨床開発段階に移行する前に、潜在的な問題を予測しなければなりません。
候補化合物の臨床試験を行うには、次の条件を満たしている必要があります。
- 医薬品の安全性試験の実施基準 (GLP) の条件に従って非臨床安全性評価が行われている。
- 適切な品質管理下で製造が実施されている。
- データおよびプロセスが CTD 形式に従って文書化されており、臨床開発段階に向けた基盤が構築されている。
モデリングと予測に生物情報学的手法を使用する傾向があると同様、医薬品的性質のデザインもコンピューター上で行う傾向が高まっています。
非臨床開発のガイドラインは、主要な規制当局 (欧州、米国、日本) の間で継続的に進められている調和の対象となっており、安全性と品質に重点が置かれています。ICH では、欧州 (EMA) および米国 (FDA) の機関が公開しているものと同様の詳細なガイダンスを、製薬業界向けに定期的に発行しています。
追加資料
- European Medicines Agency (2007a). Guideline on strategies to identify and mitigate risks for first-in human trials with investigational medicinal products. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf
- European Medicines Agency (2007b). Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002989.pdf
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
- European Medicines Agency (2011). Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
参照文献
- Image reproduced from ICH (2015). M4: The Common Technical Document. Retrieved 11 July, 2021, from https://www.ich.org/page/ctd
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
A2-2.01.1-1.2