

Introduction
The non-clinical (or pre-clinical) development phase primarily aims to identify which candidate therapy has the greatest probability of success, assess its safety, and build solid scientific foundations before transition to the clinical development phase.
Also, during the non-clinical development phase, the candidate compound should meet non-medical objectives, including defining the intellectual property rights and making enough medicinal product available for clinical trials. The non-clinical development of a medicine is complex and regulatory-driven.
Basics, key definitions, and concepts
‘Non-clinical’ or ‘pre-clinical’ ?
The terms ‘non-clinical’ and ‘pre-clinical’ are often used interchangeably.
Although of critical relevance in the pre-clinical steps of the development, non-clinical studies can be performed at any time during the life-cycle of the product, much of which is better performed as early as possible in order to avoid surprises later in the development.
Beyond identification of the pharmacodynamics (what a medicine does to the body), pharmacokinetics (what the body does to the medicine), and toxicology of the candidate compound before administration in humans, the data from non-clinical studies are used to refine, consolidate, and add information to update the safety profile of the product during the pre-clinical phase, at the time of registration, and during the life-cycle of the medicinal product.
In silico, in vitro, and in vivo studies
The studies in non-clinical development are performed:
- In silico: ‘performed on computer or via computer simulation’, e.g. predicting the toxicology profile of a product using its chemical structure from data-based approaches.
- In vitro (Latin for ‘within the glass’): performing a procedure in a controlled environment outside of a living organism, e.g. use of hepatocyte (cells from the liver) cultures for metabolism studies.
- In vivo (Latin for ’within the living’): experimentation using a whole, living organism as opposed to tissues or cells, i.e. animals, humans or plants.
What are the key aspects of Chemistry, Manufacturing, Control (CMC) during non-clinical development?
All non-clinical development studies necessitate manufacture of an adequate active substance:
- Small quantities (milligrams to grams) are usually needed for non-clinical studies; a scale-up process must then be developed to produce larger quantities for clinical trials and later on, after approval, for the market
- For Good Laboratory Practice (GLP) studies, qualified or Good Manufacturing Practice (GMP) batches of the active substance are required.
Some key CMC steps during the non-clinical development phase include:
- Determining the dose and administration
- Detailed physico-chemical characterisation
- Stability testing & impurity analysis
- Developing and validating methods to quantify the active substance in body fluids like blood, plasma, urine in activity and side effect studies
- Develop a prototype for what will be used in the clinic.
The non-clinical development process
Non-clinical development activities parallel the research activities. They should support the planned clinical development programme by addressing the objectives and questions outlined below.
Objectives
Once a candidate compound is identified, the non-clinical development should start answering the following questions, and answers will come from specific assessments/studies:
- Does it work? → efficacy assessment
- How will it be delivered and how will the body react? → profiling
- Is it safe? → toxicology/safety
- Is the manufacture viable and controllable?
Non-clinical development activities can continue throughout the life-cycle of the product, although the earlier these questions are answered, the easier it is to identify the profile of the patient who will benefit most.
Project management
The non-clinical development programme is complex and requires solid project management and communication skills in driving multidisciplinary teams. The project team needs to understand the intended clinical plan in order to define the non-clinical plan and related activities.
The profile provides a framework to execute the non-clinical development strategy, defining goals, risk, liabilities, metrics and Go/No-Go decision-making. Profile implementation helps to keep the focus of the project on key product criteria, to help ‘Go/No-Go’ decisions in a timely manner and to reduce the overall project risk (i.e., continued development of a non-useful product).
Non-clinical regulatory guidelines
There are many players involved in the development of medicines, and each organisation or institution follows their own set of rules. For instance, companies have their Standard Operating Procedures (SOP). In addition to Good Clinical Practice provisions, guidelines can be consulted at the European Medicines Agency (EMA) website.
- They are either general or more specific addressing scientific and technical aspects (e.g. specific to required toxicology studies).
- They must be strictly followed for any new marketing authorisation application; any deviation must be justified.
Data are presented according to the Common Technical Document (CTD) format as defined by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). The agreement to assemble all the Quality, Safety, and Efficacy information in this common format (the CTD) has revolutionised the regulatory review process, and has led to harmonised electronic submissions that, in turn, enable the implementation of good review practices. For industry, it has eliminated the need to reformat the information for submission to the different regulatory authorities (the ICH brings together the regulatory authorities and pharmaceutical industry of Europe, Japan, and the US to discuss scientific and technical aspects of medicines registration).
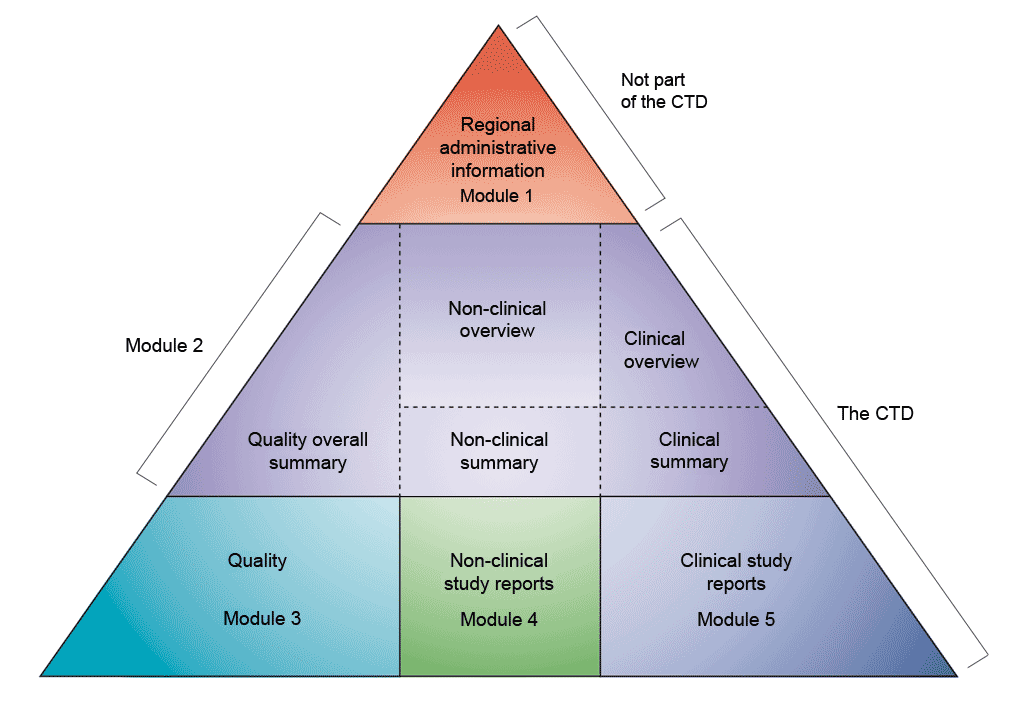
The CTD is organised into five modules (see figure above). In July 2003, the CTD became the mandatory format for new marketing authorisation applications in the EU and Japan, and the strongly recommended format of choice for New Drug Applications (NDA) submitted to the US Food and Drug Administration (FDA).
Summary
The non-clinical development phase is critical and must anticipate potential problems before a compound moves into the clinical development phase.
Admission of a candidate compound to clinical studies requires:
- Non-clinical safety evaluation obtained under Good Laboratory Practices (GLP) conditions.
- Manufacturing executed under proper quality control.
- Data and process documented according to the CTD format and building foundations for the clinical development phase.
There is an increased tendency to design medicine-like properties in silico, as well as to use bioinformatics methods for modelling and prediction.
Guidelines for non-clinical development are the subject of continuous harmonisation between the main regulatory authorities (Europe, US and Japan) highlighting safety and quality: ICH regularly issues detailed guidance for the pharmaceutical industry similar to those published by European (EMA) and US (FDA) agencies.
Further Resources
- European Medicines Agency (2007a). Guideline on strategies to identify and mitigate risks for first-in human clinical trials with investigational medicinal products. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf
- European Medicines Agency (2007b). Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002989.pdf
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
- European Medicines Agency (2011). Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
References
- Image reproduced from ICH (2015). M4: The Common Technical Document. Retrieved 11 July, 2021, from https://www.ich.org/page/ctd
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
A2-2.01.1-1.2