Last update: 15 juli 2015


Introduktion
Den prækliniske udviklingsfase har primært til formål at identificere, hvilken kandidatbehandling der med størst sandsynlighed vil virke, vurdere dens sikkerhed og opbygge et solidt videnskabeligt fundament før overgangen til den kliniske udviklingsfase.
Under den prækliniske udviklingsfase skal kandidatstoffet desuden opfylde ikke-medicinske krav, bl.a. at definere de intellektuelle rettigheder og sikre, at en tilstrækkelig mængde af lægemidlet er tilgængelig til kliniske forsøg. Et lægemiddels prækliniske udvikling er kompleks og drevet af lovmæssig regulering.
Grundlæggende principper, vigtige definitioner og begreber
“Non-klinisk” eller præklinisk”?
Begreberne “non-klinisk” og “præklinisk” bruges ofte i flæng.
Selvom non-kliniske undersøgelser har afgørende relevans i de prækliniske udviklingstrin, kan de udføres når som helst i løbet af produktets livscyklus. Det er en fordel at udføre dem så tidligt som muligt for at undgå overraskelser senere i udviklingen.
Ud over identifikation af kandidatlægemidlets farmakodynamikken (det, som et lægemiddel gør ved kroppen), farmakokinetik (det, som kroppen gør ved lægemidlet) og toksikologi (giftig effekt) før indgivelse i mennesker bruges dataene fra non-kliniske undersøgelser til at finjustere, konsolidere og tilføje oplysninger for at opdatere produktets sikkerhedsprofil i løbet af den prækliniske fase, på registreringstidspunktet og i løbet af lægemidlets livscyklus.
In silico-, in vitro– og in vivo-undersøgelser
Undersøgelserne i non-klinisk udvikling udføres:
- In silico: “udføres på computer eller via computersimulation”, f.eks. for at forudsige et produkts toksikologiprofil ved hjælp af dets kemiske struktur ud fra databaserede fremgangsmåder.
- In vitro (latinsk: “i glasset”): at udføre en procedure i et kontrolleret miljø uden for en levende organisme, f.eks. ved brug af hepatocytdyrkninger (celler fra leveren) til metabolismeundersøgelser.
- In vivo (latinsk: “i det levende”): eksperiment med en hel, levende organisme i modsætning til væv eller celler, dvs. dyr, mennesker eller planter.
Hvad er de vigtigste aspekter af kemi, produktion, kontrol (Chemistry, Manufacturing, Control – CMC) under non-klinisk udvikling?
Alle non-kliniske udviklingsundersøgelser nødvendiggør produktionen af et tilstrækkeligt aktivt stof:
- Der skal normalt bruges små mængder (milligram til gram) til non-kliniske undersøgelser. Der skal derefter udvikles en opskaleringsproces for at producere større mængder til kliniske forsøg og senere – efter godkendelsen – til markedet
- I forbindelse med undersøgelser efter god laboratoriepraksis (GLP) kræves der partier af det aktive stof, der er fremstillet efter god fremstillingspraksis (GMP).
Nogle af de vigtigste trin i kemisk produktion og kontrol (CMC) i løbet af den prækliniske udviklingsfase er:
- Fastlæggelse af dosen og indgivelsen
- Detaljeret fysisk-kemisk karakterisering
- Stabilitetstest og urenhedsanalyse
- Udvikling og validering af metoder til at kvantificere det aktive stof i kropsvæsker som blod, plasma og urin i aktivitets- og bivirkningsundersøgelser
- Udvikl en prototype til det, der skal bruges på klinikken.
Den prækliniske udviklingsproces
Prækliniske udviklingsaktiviteter kører sideløbende med forskningsaktiviteterne. De skal understøtte det planlagte kliniske udviklingsprogram ved at adressere formålene og spørgsmålene, der er angivet nedenfor.
Formål
Når et kandidatstof er blevet identificeret, skal den prækliniske udvikling begynde at besvare følgende spørgsmål, og svarene kommer fra specifikke vurderinger/undersøgelser:
- Virker det? → vurdering af effekt
- Hvordan bliver det indgivet, og hvordan reagerer kroppen? → profilering
- Er det sikkert? → toksikologi/sikkerhed
- Er fremstillingen funktionsdygtig og mulig at kontrollere?
Prækliniske udviklingsaktiviteter kan fortsætte gennem hele produktets livscyklus. Jo tidligere disse spørgsmål besvares, desto lettere er det at identificere profilen for den patient, der vil drage størst fordel af lægemidlet.
Projektledelse
Det prækliniske udviklingsprogram er komplekst og kræver solide projektledelses- og kommunikationsfærdigheder med henblik på at styre tværfaglige teams. Projektteamet skal forstå den tilsigtede kliniske plan for at kunne definere den prækliniske plan og relaterede aktiviteter.
Profilen indeholder en struktur til at eksekvere den prækliniske udviklingsstrategi og definere mål, risici, ulemper, målinger og beslutninger om at fortsætte eller ej (Go/No-Go). Profilimplementeringen hjælper med at holde fokus i projektet på de vigtige produktkriterier, hjælpe med at træffe rettidige beslutninger og reducere den overordnede risiko i projektet (dvs. fortsat udvikling af et produkt, der ikke har den ønskede effekt).
Prækliniske regulatoriske retningslinjer
Der er mange aktører involveret i udviklingen af lægemidler, og hver enkelt organisation eller institution følger sit eget regelsæt. Virksomheder har f.eks. deres operative standardprocedurer (SOP). Ud over bestemmelser for god klinisk praksis kan der findes retningslinjer på Det Europæiske Lægemiddelagenturs (EMA) websted.
- De er enten generelle eller mere specifikke og adresserer videnskabelige og tekniske aspekter (f.eks. specifikt i forhold til påkrævede toksikologiundersøgelser).
- De skal følges minutiøst for enhver ny ansøgning om markedsføringstilladelse. Der skal redegøres for enhver afvigelse.
Data præsenteres i henhold til formatet for et fælles teknisk dokument (CTD – Common Technical Document), som er defineret af det internationale råd for harmonisering (ICH) til tekniske krav i forbindelse med registrering af lægemidler til brug i mennesker. Beslutningen om at samle alle oplysninger om kvalitet, sikkerhed og virkning i dette fælles format (CTD) har revolutioneret processen til indsendelse af godkendelsesansøgninger og har ført til harmoniserede elektroniske indsendelser, der efterfølgende gør det muligt at implementere gode godkendelsesprocedurer. For virksomheder har det elimineret behovet for at omformatere de oplysninger, som indsendes til de forskellige lægemiddelmyndigheder (ICH samler lægemiddelmyndighederne og medicinalindustrien i Europa, Japan og USA for at gennemgå de videnskabelige og tekniske aspekter af lægemiddelregistrering).
-
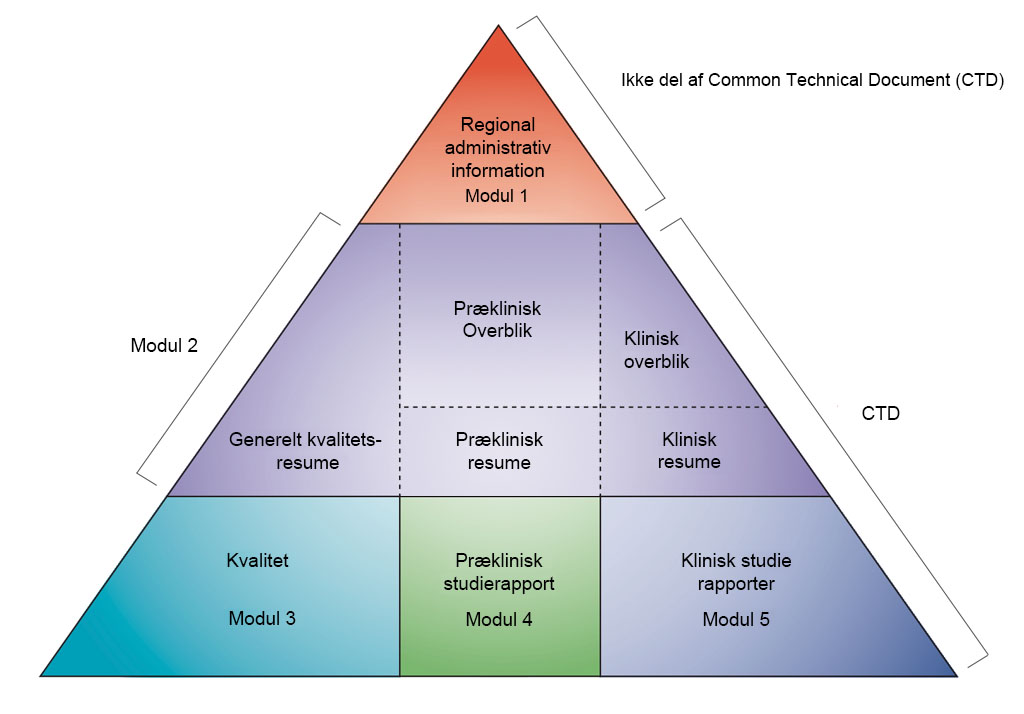
- Non-clinical development in CTD modules. Adapted from ICH CTD (see reference 1)
CTD'en er opdelt i fem moduler (se figuren ovenfor). I juli 2003 blev CTD'en det obligatoriske format for nye ansøgninger om markedsføringstilladelse i EU og Japan, og det er det foretrukne format, der kraftigt anbefales til ansøgninger om nye lægemidler, som sendes til den amerikanske Food and Drug Administration (FDA).
Sammenfatning
Den prækliniske udviklingsfase er ekstremt vigtig og skal imødegå potentielle problemer, før et lægemiddel går ind i den kliniske udviklingsfase.
Anvendelse af et kandidatstof i kliniske undersøgelser kræver følgende:
- Præklinisk sikkerhedsevaluering, der indhentes i henhold til god laboratoriepraksis (GLP).
- Fremstilling udført under grundig kvalitetskontrol.
- Data og proces dokumenteret i henhold til CTD-format og opbygning af fundamentet til den kliniske udviklingsfase.
Der er en stigende tendens til at designe lægemiddellignende karakteristika in silico og bruge bioinformatikmetoder til modellering og forudsigelser.
Retningslinjer for præklinisk udvikling er genstand for kontinuerlig harmonisering mellem de primære lægemiddelmyndigheder (Europa, USA og Japan), og de sætter fokus på sikkerhed og kvalitet: ICH udsteder med jævne mellemrum detaljerede vejledninger til medicinalindustrien i stil med dem, der udgives af europæiske (EMA) og amerikanske (FDA) styrelser.
Flere ressourcer
- European Medicines Agency (2007a). Guideline on strategies to identify and mitigate risks for first-in human trials with investigational medicinal products. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf
- European Medicines Agency (2007b). Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002989.pdf
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
- European Medicines Agency (2011). Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
Referencer
- Image reproduced from ICH (2015). M4: The Common Technical Document. Retrieved 11 July, 2021, from https://www.ich.org/page/ctd
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
A2-2.01.1-1.2