Last update: 23 november 2015


Introduktion
I det traditionelle paradigme for kliniske forsøgsdesign skal hver ny behandling gennemgå en nøje udviklingsproces. Når fase I-forsøgene er gennemført, er det nødvendigt med et fase II-forsøg for at påvise tilstrækkelig virkning og sikkerhed. Når dette er blevet påvist, overgår lægemidlet til fase III-forsøgene, hvor det sammenlignes med en standardbehandling (kontrolbehandling). At gøre dette separat for hver behandling kræver lang tid, mange patienter og betydelige finansielle ressourcer. Desuden er der med den traditionelle tilgang ikke mulighed for ændringer i løbet af forsøget.
En ny tilgang til kliniske forsøgsdesign er et adaptivt klinisk forsøgsdesign. I adaptive kliniske forsøg er det planlagt på forhånd, at der er mulighed for at ændre et eller flere fastlagte aspekter af forsøget. Dette baseres som regel på analysen af interim-data fra deltagerne under forsøget.
Der er mange grunde til at bruge adaptive design (eller adaptive forløb) i kliniske forsøg. I et miljø, der er præget af økonomiske udfordringer, kan adaptive design virke fordelagtige for medicinalindustrien, akademiske institutioner, klinikere og patienter.
Adaptive design
Adaptive design er relativt fleksible kliniske forsøgsdesign, som giver mulighed for ændringer i løbet af forsøget for at strømline og optimere processen. Der foretages analyser af de indsamlede forsøgsdata på bestemte tidspunkter i forsøget. De kan blive foretaget på en fuldstændig blindet eller ublindet måde, og de kan finde sted med eller uden formel statistisk hypoteseafprøvning. Det er vigtigt, at processen kun ændres på en sådan måde, at forsøgets validitet og integritet ikke berøres.
Adaptive design kan give operationelle udfordringer på grund af deres kompleksitet, og processen med at tilpasse et forsøg kan medføre skævheder (bias). Denne bias kan være statistisk eller operationel – f.eks. hvis en tilpasning viser, at resultaterne af et forsøg peger i en bestemt retning.
Det adaptive design kan forbedre forsøgseffektiviteten for sponsoren og forsøgsdeltagerne. Hvis et sådant forsøg ikke udføres korrekt, er der dog høj risiko for, at det kan generere kliniske resultater, som er vanskelige at fortolke eller oversætte til daglig praksis.
Adaptive design ved sjældne sygdomme
Kliniske forsøg i forbindelse med sjældne sygdomme er typisk små af nødvendighed. Planlægning af et lille klinisk forsøg, navnlig når der er tale om en sjælden sygdom, kan være forbundet med adskillige udfordringer. Små forsøg udviser mere variation end større forsøg, hvilket betyder, at standarddesign kan føre til forsøg, der kun er hensigtsmæssige for store effekter.
De specifikke krav til forsøg med sjældne sygdomme gør adaptive design særligt fordelagtige. Klassiske forsøg med sjældne sygdomme har typisk en styrke, der er beregnet på store effekter. En statistisk tests styrke er testens evne til at vise en effekt, hvis en sådan effekt reelt eksisterer. Statistisk set er det sandsynligheden for, at den på korrekt vis vil føre til, at nulhypotesen forkastes.
I nogle tilfælde er vi måske ikke i stand til at forkaste nulhypotesen, ikke fordi den er sand, men fordi vi ikke har tilstrækkelig evidens imod den. Dette kan skyldes, at forsøget ikke er stort nok til at forkaste nulhypotesen. Som sådan kan en tests styrke beskrives som sandsynligheden for ikke at lave en type II-fejl (ikke at forkaste nulhypotesen, når den rent faktisk er falsk).
Adaptive design udgør et godt alternativ, fordi:
- De afkorter udviklingsprocessen, uden at det går ud over validiteten eller virkningen
- Ineffektive behandlinger kan identificeres tidligere
- De muliggør en mere effektiv brug af ressourcer.
Det er dog vigtigt at erkende, hvad et adaptivt design kan eller ikke kan gøre, når det gælder sjældne sygdomme. Det vigtigste er, at adaptive design ikke kan gøre et lægemiddel mere effektivt. De kan dog identificere ineffektive behandlinger tidligere. En sådan tidlig identifikation kan minimere de ressourcer, der tildeles undersøgelsen af en ineffektiv behandling, og gør det muligt at omfordele ressourcerne til mere lovende behandlinger.
Mulige tilgange i adaptivt design
Begrebet “adaptivt” dækker en række forskellige design, men de fleste af dem følger en simpel struktur. I et adaptivt forsøg er der lærings- og bekræftelsesstadier, som følger en tilgang, der minder om den overordnede kliniske udviklingsproces over flere forsøgsprotokoller (fase I, fase II og fase III). Som et resultat heraf kan der foretages ændringer af hypoteserne eller designparametrene.
Læringsstadier:
- Større designelementer kan ændres (f.eks. droppe behandlingsarme)
- Statistisk usikkerhed (f.eks. bias, variation eller forkert udvælgelse)
- Estimering af behandlingseffekterne (positive eller negative)
Bekræftelsesstadier:
- Kontrol af statistiske fejl og operationel bias er af yderste vigtighed
- Stærk kontrol af type I-fejl er påkrævet (f.eks. at finde en behandling effektiv, når den reelt ikke er det).
Det mest almindeligt anvendte adaptive design er forsøg med tidlige stop-regler på grund af futilitet (når behandlingen eller forsøget ikke viser nogen nyttige resultater) eller virkning.
Disse regler er opstillet på forhånd og verificeres ved hjælp af en eller flere interim-analyser. De forhindrer deltagerne i at tage lægemidler, som ikke ville have en gavnlig effekt, eller som ikke er sikre. Vigtigst af alt er, at hvis det konstateres, at forsøgslægemidlet er klinisk mere effektivt end kontrollægemidlet, ville det være uetisk at fortsætte med at administrere det mindre effektive kontrollægemiddel. Tidlige stop-regler på grund af futilitet gør det muligt at indstille administrationen af et mindre effektivt kontrollægemiddel.
Der er også design, hvor behandlingsarme droppes i løbet af forsøget, eller hvor en underpopulation vælges på baggrund af en interessant biomarkør.
Nogle design gør det muligt at re-estimere stikprøvestørrelsen, f.eks. en forøgelse af patientpopulationen, hvis resultaterne ser lovende ud.
Adaptiv randomisering er et andet eksempel på et intuitivt tiltalende design. Med dette design vil en større andel af patienterne blive behandlet med den “bedre” arm (hvis en sådan forefindes). Disse adaptive forsøgsdesign er for det meste baseret på ublindede interim-analyser til vurdering af behandlingernes effekt – hvilket betyder, at analytikerne er klar over, hvilken behandling deltagerne er blevet tildelt.
Eksempler på adaptive forsøgsdesign
Eksempel 1: Gruppesekventielt design
Et gruppesekventielt design er et typisk eksempel på et fase III-forsøg med regler om tidlig standsning på grund af futilitet eller virkning. I det forsøgseksempel, der er vist i diagrammet nedenfor, blev patienterne randomiseret til førstevalgsbehandling med enten ét lægemiddel alene eller to lægemidler i kombination.
Der var to interim-stadier, hvor det var muligt at stoppe forsøget tidligt og foretage en analyse, inden alle forsøgsresultaterne var samlet. Forsøget kunne være blevet stoppet:
- Ved interim-stadie 1 på grund af futilitet baseret på progressionsfri overlevelse (PFS) – dvs. hvorvidt patienten undgår en specifik kræftforms progression eller ej
- Ved interim-stadie 2 på grund af futilitet eller virkning baseret på samlet overlevelse.
Gruppesekventielt design er et klassisk eksempel, der ofte bliver glemt, når man taler om adaptivt design, eftersom det allerede blev anvendt, før andre adaptive design blev mere almindelige. Tilpasningsmulighederne planlægges i forvejen i forsøgsdesignet, hvilket betyder, at styrke og type I-fejl eller sekventielle test er relativt lette at justere, når der foretages flere test. Hermed bevares den overordnede styrke og type I-fejl.
-
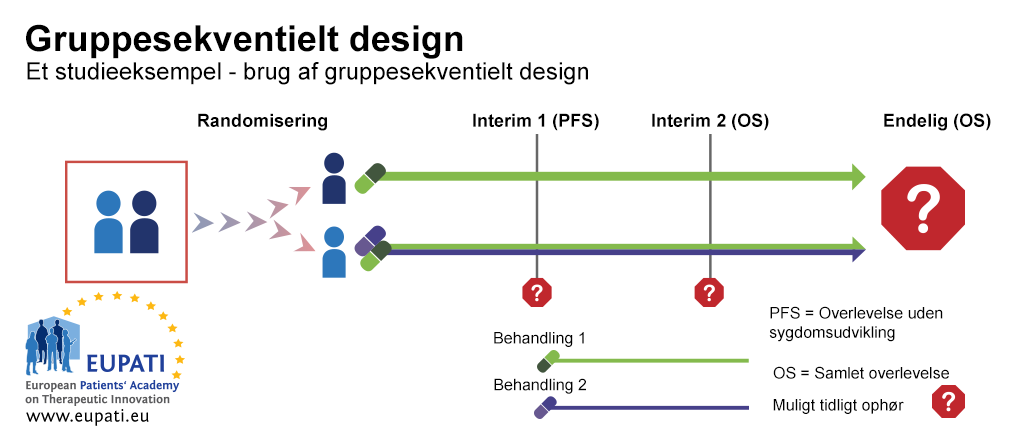
- Gruppesekventielt design giver mulighed for tidlig standsning på baggrund af progressionsfri overlevelse eller samlet overlevelse. I dette eksempel blev deltagerne randomiseret til en af de to arme og fik enten behandling 1 eller en kombination af behandling 1 og behandling 2.
Eksempel 2: Flertrinsdesign med flere behandlingsarme (MAMS)
Flertrinsforsøg med flere behandlingsarme (MAMS) er et nyt paradigme til udførelse af randomiserede, kontrollerede forsøg, der anvender et interessant adaptivt design.
MAMS-forsøg giver mulighed for en samtidig vurdering af en række forsøgsbehandlinger mod én enkelt kontrolarm. MAMS-forsøg giver tidligere svar og er potentielt mere omkostningseffektive end en række traditionelt designede forsøg.
I dette eksempel ser vi et design, hvor der anvendes flere arme og flere trin på samme tid.
MAMS-designet kræver et endeligt primært og et mellemliggende primært resultatmål. Det endelige resultatmål er det, som de endelige konklusioner skal baseres på, mens det mellemliggende resultatmål udgør et middel til at screene for ny evidens.
Ved den første interim-analyse i eksemplet ovenfor anses det nye regime 2 for ikke at have tilstrækkelige fordele sammenlignet med kontrolregimet, og det fortsætter ikke til stadie 2. Ved den anden interim-analyse stoppes rekrutteringen til de nye regimer 1 og 4, og kun kontrolregimet og det nye regime 3 fortsætter til forsøgets afslutning og overgår til fase III-undersøgelserne.
Fordele ved MAMS-design:
- Færre patienter Med dette design udføres der flere forsøg på én gang, hvilket er med til at reducere antallet af patienter, som randomiseres til kontrolarmen
- Mindre samlet tidsforbrug påkrævet til lægemiddelforskning De mellemliggende trin i MAMS-designet erstatter det separate fase II-trin. Beslutningen om, hvorvidt lægemidlet er tilstrækkelig aktivt, indarbejdes som en pilotfase i dette forsøg.
- Færre ansøgninger og godkendelser påkrævet Det regulatoriske arbejde bliver gjort for ét forsøg i stedet for flere forsøg.
- Fleksibelt Uinteressante arme kan droppes, og nye arme kan tilføjes.
- Reducerede omkostninger Dette forsøgsdesign kræver færre patienter, færre regulatoriske ansøgninger og et mindre samlet tidsforbrug, hvilket alt sammen er med til at nedbringe udviklingsomkostningerne.
Ulemper ved MAMS-design:
- Gennemførelseskarakteristika Da denne tilgang er kompleks, kan den være vanskelig at administrere, og den kræver en lang række simuleringer under designprocessen.
- Påkrævet antal patienter
Dette afhænger af gennemførelseskarakteristikaene, men hvis der tilføjes behandlingsarme i løbet af forsøget, kan det være vanskeligt at forudsige budget- og regulatoriske problemer.
- Forsøgets varighed
Hvis der tilføjes behandlingsarme, bliver det vanskeligt at forudsige, hvornår forsøget stoppes.
- Fortsat tilvækst (rekruttering) i kontrolarmen For at undgå en tidsmæssig skævhed, når der tilføjes nye behandlingsarme, skal rekrutteringen til kontrolarmen fortsætte under hele forsøget. Det skal også overvejes, hvad der sker, hvis en ny standardbehandling bliver tilgængelig i løbet af forsøget – er kontrollen så stadig relevant?
- Sammenligning mellem forsøgsarmene MAMS-designet giver kun mulighed for sammenligninger mellem individuelle behandlingsarme og kontrolarmen. Det giver ikke mulighed for at sammenligne de individuelle behandlingsarme med hinanden.
Eksempel 3: Sammenhængende fase II-/fase III-design
Sammenhængende fase II-/fase III-design anvendes ofte ved sjældne sygdomme og kaldes også for et "kombinationsforsøg". I eksemplet nedenfor randomiseres patienterne til tre behandlingsarme i det første stadie af designet (fase IIb). Den første behandlingsarm er kontrolarmen, hvor patienterne får standardbehandlingen. Patienterne i den anden og tredje behandlingsarm får forskellige behandlinger, nemlig behandling 1 eller behandling 2.
Ved afslutningen af det første stadie (fase IIb) sammenlignes behandling 1 og behandling 2 ud fra den bedste progressionsfri overlevelse (PFS). Den mindst effektive behandlingsarm droppes. Den anden behandlingsarm fortsætter i det næste stadie (fase III). I dette stadie foretages der en virkningssammenligning med standardbehandlingen.
Fordele ved sammenhængende fase II-/fase III-design
- Er med til at mindske bias Begge trin gennemføres uafhængigt af hinanden, og resultaterne af begge trin kombineres til sidst i et samlet testresultat.
- Afkorter varighed og patienteksponering
Fase II og fase III gennemføres inden for rammerne af ét forsøg.
- Relativt fleksibel Den måde, hvorpå behandlingsarmen til den endelige sammenligning vælges i fase II-delen og kombineres med fase III-delen, er relativt fleksibel.
- Effektiv anvendelse af ressourcer
Patienter fra både fase II og fase III bidrager med data til de endelige resultater.
Ulemper
- Komplicerede statistiske analyser
Dette design kræver statistiske aspekter, som ikke er så ligetil.
- Rekrutteringshuller Der er et hul i rekrutteringen mellem de to faser, mens man venter på, at der er samlet tilstrækkelige data til at foretage interim-analysen, som afgør, om man skal fortsætte eller ej.
- Logistiske udfordringer Dette design er logistisk udfordrende – det kræver et hurtigt dataflow, så der kan følges op på antallet af hændelser i analysen.
- Vanskeligheder som følge af langsigtede endepunkter Dette design kræver, at informationen om progressionsfri overlevelse er tilgængelig relativt hurtigt. Dette bliver vanskeligere, når endepunkterne er langsigtede.
- Risiko for tabt information Ved at kombinere to arme risikerer man, at information går tabt
Patientinddragelse
Patientinput til adaptivt design kan hjælpe forskerne med at identificere det mest velegnede design ved at bidrage til at definere og forstå patientpopulationens behov og krav. Patienterne kan også blive inddraget i datasikkerhedsmonitoreringskomitéen.
Konklusioner
Nye forsøgsdesign kan muliggøre:
- Fleksible designstrategier
- Mere effektiv anvendelse af ressourcerne
- Kortere udviklingsproces
Ud fra et regulatorisk perspektiv er det vigtigt at bevare validiteten og integriteten af adaptive design i kliniske forsøg:
- Adressér det samme spørgsmål som i det klassiske forsøgsdesign
- Kontrollér operationel bias
- Kontrollér eventuelle statistisk signifikante fejl
- Fortolkning af resultater
Bilag
A2-4.08-v1.2