

La galenica è il processo che trasforma un principio attivo in un farmaco pronto per l’uso, in grado di essere dosato nel modo richiesto. La formulazione galenica si occupa dei principi della preparazione e composizione dei farmaci al fine di ottimizzare il loro assorbimento e fa parte della farmaceutica, la disciplina (o scienza) che studia lo sviluppo delle forme di dosaggio.
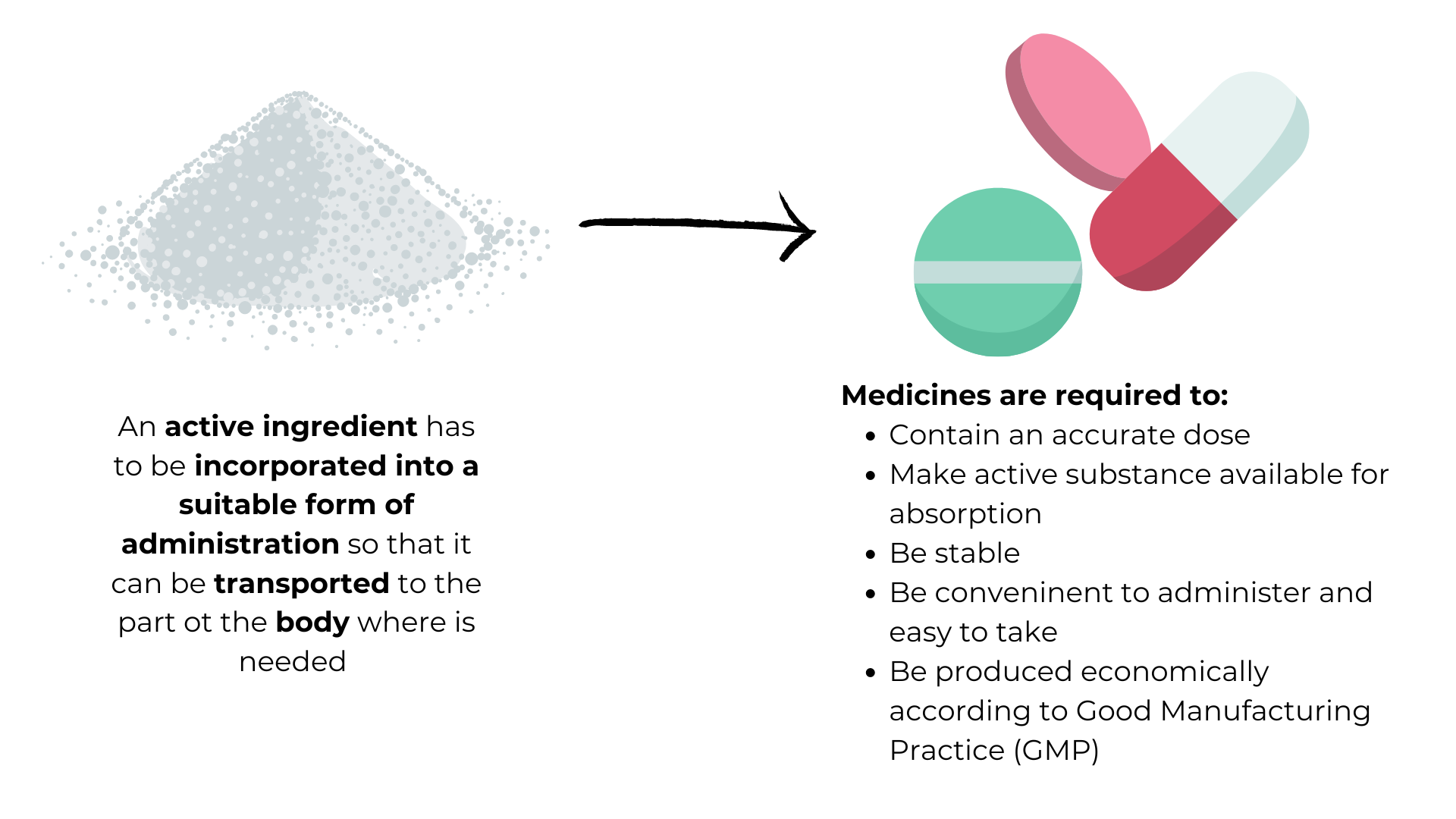 Per lo sviluppo della formulazione, gli studi di valutazione della sicurezza e gli studi clinici è necessario un principio attivo. In un processo di sviluppo chimico, viene prodotto un quantitativo sufficiente di principio attivo di qualità affinché sia utilizzato in studi di sicurezza e gli scienziati formulino un prodotto medicinale.
Per lo sviluppo della formulazione, gli studi di valutazione della sicurezza e gli studi clinici è necessario un principio attivo. In un processo di sviluppo chimico, viene prodotto un quantitativo sufficiente di principio attivo di qualità affinché sia utilizzato in studi di sicurezza e gli scienziati formulino un prodotto medicinale.
Il prodotto medicinale comprenderà la formulazione del principio attivo (compressa, crema, sospensione,soluzione), eccipienti (sostanze inattive come il lattosio) e il dispositivo d’imballaggio/erogazione (confezione blister, flacone, inalatore, fiala, siringa pre-riempita).
Formulazione galenica per studi preclinici
Gli studi preclinici di sicurezza hanno i seguenti obiettivi:
- Esplorare la risposta fino alle dosi massime tollerabili
- Individuare potenziali rischi
- Generare una quantità sufficiente di dati al fine di realizzare una valutazione del rischio
- Contribuire alla selezione della dose per studi clinici iniziali
- Suggerire “marcatori” al fine di monitorare la sicurezza negli esseri umani
- Fornire delle basi per sperimentazioni specializzate mirate.
In ogni caso, tali studi di sicurezza non sono in grado di garantire necessariamente la sicurezza negli esseri umani o di prevedere le risposte da parte degli stessi. L’analisi della sicurezza valuta i margini di sicurezza per dati relativi ad animali ed esseri umani. Per questi tipi di studi si svolgono le seguenti osservazioni:
- Dose somministrata
- Entità e durata dell’esposizione sistemica
- Esposizione sistemica giornaliera
- Esposizione e identità dei metaboliti
- Esposizione negli organi bersaglio.
Gli studi di sicurezza vengono svolti sul principio attivo, così come su tutte le sostanze e tutti i solventi, prodotti di degradazione, eccipienti e gli altri materiali attivi ed estratti correlati di cui la formulazione finale è costituita.
Formulazione galenica per studi clinici
I fattori da prendere in considerazione nello sviluppo di una forma idonea di somministrazione comprendono l’accettabilità da parte del paziente e requisiti specifici del principio attivo. Le sostanze inattive o eccipienti possono costituire la maggior parte del volume di un farmaco e servono da vettori del composto attivo. Amido di mais o lattosio, ad esempio, vengono utilizzati nelle compresse, mentre emulsioni acqua-olio sono usate negli unguenti.
La forma di somministrazione influenza anche l’assorbimento, la disponibilità del principio attivo e quindi l’effetto terapeutico del farmaco. Determina infatti il modo in cui il principio attivo entra nell’organismo, dove e con quale dosaggio viene rilasciato e il tempo richiesto perché venga assorbito. Inoltre, il modo di somministrazione deve assicurare che il paziente sia in grado di dosare il farmaco in maniera sicura e di maneggiarlo facilmente.
Gli scienziati che si occupano della formulazione si assicurano che la sostanza possa essere assorbita dall’organismo e che la dose terapeutica raggiunga l’organo previsto. Non tutti principi attivi sono adatti all’ingestione sotto forma di compressa e particolari esigenze riguardanti la forma di somministrazione (iniezione oculare, prodotti per inalazione oppure compresse che si dissolvono in bocca) creano regolarmente nuove sfide per tali studiosi.
Buona prassi di fabbricazione (GMP)
Tutti i medicinali, compresi i farmaci generici e i biosimilari, devono essere prodotti secondo una prassi di buona fabbricazione (GMP, Good Manufacturing Practice).
Queste linee guida forniscono direttive per la fabbricazione, le analisi e le garanzie di sicurezza, al fine di assicurare che un farmaco sia sicuro per il consumo umano. Molti paesi hanno specifiche leggi e regolamenti, i quali impongono che i produttori di dispositivi farmaceutici e medici seguano procedure di GMP. La maggioranza dei regolamenti si basa su linee guida concordate a livello internazionale.
Le linee guida di GMP seguono alcuni principi di base:
- Igiene: le strutture di fabbricazione di prodotti farmaceutici devono mantenere gli ambienti puliti e in condizioni igieniche adeguate.
- Condizioni ambientali controllate, al fine di prevenire contaminazioni incrociate di un prodotto medicinale causate da altri composti o da particolato atmosferico estraneo, i quali potrebbero rendere il farmaco pericoloso per il consumo umano.
- Le procedure di fabbricazione sono chiaramente definite e controllate:
- Tutte le operazioni critiche vengono convalidate per assicurare stabilità e aderenza alle specifiche.
- Viene esaminata qualsiasi modifica al processo.
- I cambiamenti aventi un impatto sulla qualità del farmaco vengono convalidati secondo necessità.
- Le istruzioni e le procedure sono scritte con un linguaggio chiaro e non ambiguo (Buona prassi di documentazione (GDP, Good Documentation Practices)).
- Gli operatori sono addestrati all’esecuzione e alla documentazione delle procedure.
- Durante la fabbricazionevengono creati registri, manualmente o mediante strumenti, per dimostrare che siano stati svolti tutti i passaggi richiesti da procedure e istruzioni definite e che la quantità e la qualità del farmaco siano quelle attese.
- Eventuali difformità vengono esaminate e documentate.
- I registri di fabbricazione (inclusa la distribuzione), che permettono di ritracciare l’intera storia di un lotto, vengono conservati in forma comprensibile e accessibile.
- La distribuzione dei farmaci riduce al minimo eventuali rischi relativi alla loro qualità.
- È disponibile un sistema per rintracciare qualsiasi lotto di medicinali dalla vendita o dalle scorte.
- Le lamentele relative ai farmaci immessi in commercio vengono prese in esame, vengono studiate le cause dei difetti qualitativi e adottate misure adeguate riguardo ai medicinali difettosi, al fine di prevenire che si ripetano episodi simili.
Risorse aggiuntive
- Linee guida di buona prassi di fabbricazione dell’UE
- Linee guida di buona prassi di fabbricazione dell’OMS (Organizzazione mondiale della sanità)
A2-2.06-V1.4