Last update: 19 november 2015


Inleiding
Dankzij klinisch onderzoek komen nieuwe geneesmiddelen en betere behandelingen beschikbaar voor patiënten. De informatie uit klinische onderzoeken over de werkzaamheid en veiligheid van deze behandelingen is belangrijk voor patiënten en hun artsen om goed geïnformeerde behandelbesluiten te kunnen nemen. Het nut van een behandeling moet op wereldschaal worden beoordeeld, rekening houdend met alle beschikbare resultaten van klinische onderzoeken naar de behandeling. Toegang tot informatie van klinische onderzoeken is een goede manier om de efficiëntie van onderzoek te verbeteren doordat het dubbel of herhaald onderzoekswerk vermindert. De transparantie van informatie uit klinisch onderzoek is belangrijk voor het vertrouwen in onderzoeksresultaten. De lezer dient de gepubliceerde informatie uit klinische onderzoeken kritisch te evalueren.
Wat zijn onderzoeksresultaten?
De resultaten van een klinisch onderzoek zijn alle gegevens, maten en statistische analyses die gedurende dat klinische onderzoek zijn voortgebracht.
Onderzoeksresultaten omvatten de volgende elementen:
- Beschrijving van de onderzoekspopulatie: Het aantal deelnemers per behandelarm dat met het onderzoek is gestart, het onderzoek heeft afgerond en voortijdig is gestopt.
- Baselinegegevens: Gegevens die aan het begin van een klinisch onderzoek worden verzameld. Deze gegevens omvatten onder meer demografische gegevens (zoals leeftijd en geslacht), kenmerken van patiënten (zoals lengte, gewicht, bloeddruk) en metingen specifiek voor het onderzoek (zoals ziektekenmerken of eerdere behandeling).
- Maten die het effect van de behandeling op deelnemers weergeven: Bijvoorbeeld activiteit van een geneesmiddel in een klinisch Fase II-onderzoek, overlevingscijfers van patiënten en/of kwaliteit van leven in klinische Fase III-onderzoeken.
- Bijwerkingen die de onderzoeksdeelnemers ondervonden: Bijvoorbeeld pijn, misselijkheid en andere bijwerkingen.
Het ‘Clinical Study Report’ (Klinisch Onderzoeksrapport, CSR) is het formele document waarin de resultaten van een klinisch onderzoek staan beschreven en dat bewijs levert voor gebruik van het geneesmiddel bij mensen. De structuur van CSR’s is vastgelegd door de toezichthoudende instanties. Het CSR wordt door de sponsor van het onderzoek opgesteld en maakt deel uit van het ‘Common Technical Document’ (Gemeenschappelijk Technisch Document, CTD). Toegang tot klinische onderzoeksrapporten (‘Clinical Study Reports’) is doorgaans beperkt tot de sponsor en de toezichthoudende instanties die een aanvraag voor een handelsvergunning beoordelen, vanwege de vertrouwelijkheid en commerciële aspecten.
Publicatie van klinische onderzoeksresultaten
Aan het einde van het klinische onderzoek en de analyse ervan, kunnen onderzoekers hun conclusies presenteren op wetenschappelijke bijeenkomsten en in medische tijdschriften. Voorafgaand aan publicatie in medische tijdschriften wordt het manuscript beoordeeld door onafhankelijke deskundigen (‘peer-review’) die door de redacteur van het tijdschrift worden aangewezen.
Publicaties moeten voldoende gegevens bevatten om de lezer in staat te stellen een eigen oordeel te vormen over de bevindingen van het onderzoek. De kwaliteit van de publicatie heeft invloed op het vertrouwen van de lezer in de betrouwbaarheid van de resultaten. Daarom zijn er verschillende richtlijnen en controlelijsten voor de rapportage van resultaten op een gestandaardiseerde wijze, afhankelijk van het soort onderzoek dat is uitgevoerd.
Op dit moment wordt door diverse organisaties gewerkt aan projecten die de registratie en openbaarmaking van informatie uit klinisch onderzoek moeten stimuleren of verplichten. In Europa verzamelt EudraCT, de Europese databank voor klinische studies van het Europees Geneesmiddelenbureau (EMA), informatie over alle klinische onderzoeken die in Europa worden uitgevoerd. Sinds juli 2014 zijn via deze databank ook samenvattingen van onderzoeksresultaten openbaar toegankelijk voor het publiek. Voor onderzoeken in de EU die zijn gestart na 1januari2015 moeten de resultaten worden gepubliceerd – het maakt daarbij niet uit of ze positief of negatief zijn. De Wereldgezondheidsorganisatie (World Health Organisation, WHO) stelt via het eigen ‘International Clinical Trials Registry Platform’ (ICTRP) internationale regels voor registratie en rapportage op voor alle klinische onderzoeken. Het ‘clinicaltrials.gov’ register in de VS hanteert een vergelijkbare aanpak.
Mate van bewijsvoering in onderzoeksresultaten
Medische beslissingen over behandelingen zijn nu grotendeels gebaseerd op ‘evidence-based medicine’ (EBM). Bij EBM (geneeskunde op basis van de mate van bewijsvoering) wordt klinische ervaring gecombineerd met het momenteel beste bewijs uit gecontroleerde klinische studies en wetenschappelijk onderzoek om zo te komen tot de beste behandeling voor een patiënt. Informatie over de veiligheid en werkzaamheid van een behandeling is belangrijk bij EBM omdat patiënten en hun artsen zo weloverwogen behandelbesluiten kunnen nemen.
EBM is gebaseerd op kennis en evaluatie van de momenteel beste bewijzen met betrekking tot de effecten van diverse behandelvormen en gezondheidszorg in het algemeen. Het is belangrijk dat bewijs met betrekking tot een behandeling niet in slechts één publicatie wordt ondergezocht. Bij vergelijking van resultaten die afkomstig zijn van uiteenlopende bronnen, is het belangrijk om te weten dat er verschillende niveaus van bewijsvoering bestaan (zie figuur 1 hieronder). De niveaus van bewijsvoering classificeren en geven de kwaliteit weer van het onderzoek en daarmee de bewijskracht die het onderzoek biedt. Gerandomiseerde, gecontroleerde geblindeerde onderzoeken bieden de beste wetenschappelijke evidentie (bewijs) van baten en risico’s, maar deze zijn niet altijd beschikbaar. Met een meta-analyse (een op statistiek gebaseerde evaluatie waarin resultaten van uiteenlopende maar gerelateerde onderzoeken tegenover en naast elkaar worden geplaatst) wordt geprobeerd om patronen, verschillen en andere relaties tussen meerdere onderzoeken te identificeren. Een meta-analyse kan een sterkere conclusie ondersteunen dan een willekeurig individueel onderzoek, maar kan verzwakt zijn vanwege publicatievertekening (‘bias’).
-
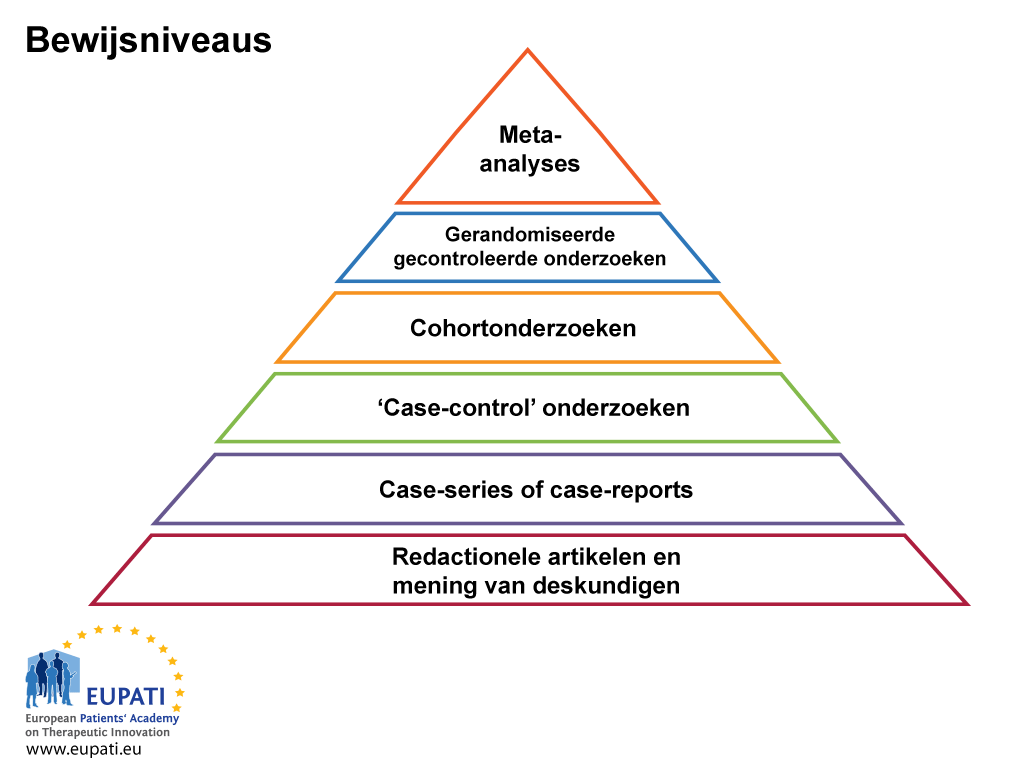
- De mate van bewijsvoering komt van pas wanneer de kwaliteit van het bewijs moet worden beoordeeld.
Over het algemeen zijn de onderzoeken:
- Gerandomiseerde onderzoeken van hoge kwaliteit met toereikend onderscheidend vermogen (‘power’) of meta-analyses van gerandomiseerde onderzoeken met statistisch consistente resultaten
- Gerandomiseerde onderzoeken met onvoldoende onderscheidend vermogen (‘power’), mogelijk vertekend (‘bias’) of met statistisch inconsistente resultaten
- Niet-gerandomiseerde onderzoeken met gelijktijdige controlegroepen
- Niet-gerandomiseerde onderzoeken met historische controlegroepen (bijvoorbeeld een gemiddeld eenarmig Fase II-onderzoek)
- Evaluatie door een comité van deskundigen, ‘case reports’, retrospectieve onderzoeken
Herkomst van fouten in publicaties
De drie meest voorkomende bronnen van fouten in publicaties zijn:1
- Het risico op misbruik en verkeerde interpretatie van statistische toetsen en hun uitkomsten, als gevolg van onduidelijkheid over de betekenis van getallen (schattingen) en de interpretatie van hypothesetoetsen (p-waarde, onderscheidend vermogen/’power’).
- ‘Data dredging’ (het zoeken naar –statistische- verbanden in gegevensverzamelingen; ook ‘data-mining’ genoemd) of het toetsen van grote aantallen hypothesen in een enkele gegevensverzameling om een positief effect te vinden. Wanneer meerdere hypothesen worden getoetst met één enkele gegevensverzameling is het bijna zeker dat sommige hypothesen onterecht statistisch significant lijken, hoewel de correlaties er in werkelijkheid niet zijn. Als onderzoekers gebruikmaken van ‘data-mining’ en niet zeer precies te werk gaan, kunnen ze gemakkelijk worden misleid door schijnbaar significante resultaten.
- Vertekening (bias). Bij onderzoek treedt vertekening op wanneer een systematische fout in de gegevensverwerking of hypothesetoets wordt geïntroduceerd door selectie of preferentie van een uitkomst of antwoord ten opzichte van andere uitkomsten of antwoorden. Vertekening (bias) is niet altijd het resultaat van doelgerichte acties – het kan ook onbedoeld worden geïntroduceerd.
Referenties
- Goldacre, B. (2010) Bad science: Quacks, hacks, and big pharma flacks. New York: Faber and Faber.
- Rennie, D., & Guyatt, G. (2002). Users’ guides to the medical literature: A manual for evidence-based clinical practice. Chicago, IL: American Medical Association.
A2-4.35.1-v1.2