Last update: 15 Ιουλίου 2015


Εισαγωγή
Η φάση της μη κλινικής (ή προκλινικής) ανάπτυξης αποσκοπεί κυρίως στον εντοπισμό της υποψήφιας θεραπείας που έχει τη μεγαλύτερη πιθανότητα επιτυχίας, στην αξιολόγηση της ασφάλειάς της και στη δημιουργία στέρεων επιστημονικών βάσεων πριν από τη μετάβαση στη φάση της κλινικής ανάπτυξης.
Επίσης, κατά τη διάρκεια της μη κλινικής φάσης ανάπτυξης, η υποψήφια ένωση θα πρέπει να πληροί μη ιατρικούς στόχους, συμπεριλαμβανομένου του καθορισμού των δικαιωμάτων πνευματικής ιδιοκτησίας και της διάθεσης επαρκούς ποσότητας φαρμάκου για κλινικές δοκιμές. Η μη κλινική ανάπτυξη ενός φαρμάκου είναι πολύπλοκη και καθορίζεται από κανονιστικές διατάξεις.
Βασικά στοιχεία, βασικοί ορισμοί και έννοιες
«Μη κλινικός» ή «προκλινικός»;
Οι όροι «μη κλινικός» και «προκλινικός» χρησιμοποιούνται συχνά εναλλακτικά.
Αν και έχουν κρίσιμη σημασία στα προκλινικά στάδια της ανάπτυξης, οι μη κλινικές μελέτες μπορούν να διεξαχθούν ανά πάσα στιγμή κατά τη διάρκεια του κύκλου ζωής του προϊόντος, μεγάλο μέρος των οποίων είναι καλύτερο να διεξάγεται όσο το δυνατόν νωρίτερα, προκειμένου να αποφεύγονται εκπλήξεις αργότερα κατά την ανάπτυξη.
Πέρα από τον προσδιορισμό της φαρμακοδυναμικής (τι κάνει ένα φάρμακο στον οργανισμό), της φαρμακοκινητικής (τι κάνει ο οργανισμός στο φάρμακο) και της τοξικολογίας της υποψήφιας ένωσης πριν από τη χορήγησή της στον άνθρωπο, τα δεδομένα από μη κλινικές μελέτες χρησιμοποιούνται για τη βελτίωση, την καθιέρωση και την προσθήκη πληροφοριών για την επικαιροποίηση του προφίλ ασφάλειας του προϊόντος κατά τη διάρκεια της προκλινικής φάσης, κατά τη στιγμή της καταχώρισης και κατά τη διάρκεια του κύκλου ζωής του φαρμάκου.
In silico, in vitro και in vivo μελέτες
Οι μελέτες στη μη κλινική ανάπτυξη διενεργούνται:
- In silico: «εκτελούνται σε υπολογιστή ή μέσω προσομοίωσης σε υπολογιστή», π.χ. πρόβλεψη του τοξικολογικού προφίλ ενός προϊόντος χρησιμοποιώντας τη χημική δομή του από προσεγγίσεις που βασίζονται σε δεδομένα.
- In vitro (λατινικά για «εντός του γυαλιού”): εκτέλεση μιας διαδικασίας σε ελεγχόμενο περιβάλλον εκτός ζώντος οργανισμού, π.χ. χρήση καλλιεργειών ηπατοκυττάρων (κύτταρα από το ήπαρ) για μελέτες μεταβολισμού.
- In vivo (λατινικά για «εντός του ζωντανού οργανισμού»): πειράματα που εκτελούνται με χρήση ολόκληρου, ζωντανού οργανισμού σε αντίθεση με ιστούς ή κύτταρα, δηλαδή ζώα, ανθρώπους ή φυτά.
Ποιες είναι οι βασικές πλευρές που συνδέονται με τη Χημεία, την Παραγωγή, τον Έλεγχο (CMC) κατά τη διάρκεια της μη κλινικής ανάπτυξης;
Όλες οι μη κλινικές μελέτες ανάπτυξης απαιτούν την παρασκευή επαρκούς δραστικής ουσίας:
- Απαιτούνται μικρές ποσότητες (χιλιοστόγραμμα έως γραμμάρια) συνήθως για μη κλινικές μελέτες- στη συνέχεια πρέπει να αναπτυχθεί μια διαδικασία κλιμάκωσης για την παραγωγή μεγαλύτερων ποσοτήτων για κλινικές δοκιμές και αργότερα, μετά την έγκριση, για την κυκλοφορία στην αγορά.
- Για μελέτες ορθής εργαστηριακής πρακτικής (GLP), απαιτούνται αναγνωρισμένες παρτίδες ή παρτίδες ορθής παρασκευαστικής πρακτικής (GMP) της δραστικής ουσίας.
Ορισμένα βασικά βήματα των διαδικασιών CMC κατά τη διάρκεια της μη κλινικής φάσης ανάπτυξης περιλαμβάνουν:
- Καθορισμό της δόσης και της χορήγησης
- Λεπτομερή φυσικοχημικό χαρακτηρισμό
- Δοκιμές σταθερότητας & ανάλυση προσμίξεων
- Ανάπτυξη και επικύρωση μεθόδων για τον ποσοτικό προσδιορισμό της δραστικής ουσίας σε σωματικά υγρά όπως αίμα, πλάσμα, ούρα σε μελέτες δραστικότητας και παρενεργειών.
- Ανάπτυξη ενός πρωτοτύπου για αυτό που θα χρησιμοποιηθεί στην κλινική.
Η μη κλινική διαδικασία ανάπτυξης
Οι δραστηριότητες μη κλινικής ανάπτυξης είναι παράλληλες με τις ερευνητικές δραστηριότητες. Θα πρέπει να υποστηρίζουν το σχεδιαζόμενο πρόγραμμα κλινικής ανάπτυξης, αντιμετωπίζοντας τους στόχους και τα ερωτήματα που περιγράφονται κατωτέρω.
Στόχοι
Μόλις εντοπιστεί μια υποψήφια ένωση, η μη κλινική ανάπτυξη θα πρέπει να αρχίσει να απαντά στα ακόλουθα ερωτήματα, και οι απαντήσεις θα προκύψουν από συγκεκριμένες αξιολογήσεις/μελέτες:
- Είναι αποτελεσματική; → αξιολόγηση της αποτελεσματικότητας
- Πώς θα χορηγηθεί και πώς θα αντιδράσει ο οργανισμός; → Δημιουργία προφίλ
- Είναι ασφαλές; → τοξικολογία/ασφάλεια
- Είναι η παραγωγή βιώσιμη και ελεγχόμενη;
Οι μη κλινικές δραστηριότητες ανάπτυξης μπορούν να συνεχιστούν καθ’ όλη τη διάρκεια του κύκλου ζωής του προϊόντος, αν και όσο νωρίτερα απαντηθούν αυτά τα ερωτήματα, τόσο ευκολότερο είναι να προσδιοριστεί το προφίλ του ασθενούς που θα ωφεληθεί περισσότερο.
Διαχείριση έργου
Το πρόγραμμα μη κλινικής ανάπτυξης είναι πολύπλοκο και απαιτεί ισχυρές δεξιότητες διαχείρισης έργων και επικοινωνίας για την καθοδήγηση διεπιστημονικών ομάδων. Η ομάδα έργου πρέπει να κατανοήσει το προβλεπόμενο κλινικό σχέδιο προκειμένου να καθορίσει το μη κλινικό σχέδιο και τις σχετικές δραστηριότητες.
Το προφίλ παρέχει ένα πλαίσιο για την εκτέλεση της στρατηγικής μη κλινικής ανάπτυξης, καθορίζοντας τους στόχους, τον κίνδυνο, τις υποχρεώσεις, τις μετρήσεις και τη λήψη της απόφασης προώθησης ή μη του προϊόντος. Η εφαρμογή του προφίλ συμβάλλει στο να παραμείνει το έργο εστιασμένο στα βασικά κριτήρια του προϊόντος, στην έγκαιρη λήψη της απόφασης προώθησης ή μη του προϊόντος και στη μείωση του συνολικού κινδύνου του έργου (π.χ. συνέχιση της ανάπτυξης ενός μη χρήσιμου προϊόντος).
Μη κλινικές κανονιστικές κατευθυντήριες γραμμές
Υπάρχουν πολλοί φορείς που εμπλέκονται στην ανάπτυξη φαρμάκων και κάθε οργανισμός ή ίδρυμα ακολουθεί τους δικούς του κανόνες. Παραδείγματος χάρη, οι εταιρείες έχουν τις δικές τους Τυποποιημένες Διαδικασίες Λειτουργίας (SOP). Εκτός από τις διατάξεις περί ορθής κλινικής πρακτικής, οι κατευθυντήριες γραμμές μπορούν να αναζητηθούν στον δικτυακό τόπο του Ευρωπαϊκού Οργανισμού Φαρμάκων (ΕΜΑ).
- Είναι είτε γενικές είτε πιο ειδικές και αφορούν επιστημονικές και τεχνικές πτυχές (π.χ. ειδικές για τις απαιτούμενες τοξικολογικές μελέτες).
- Πρέπει να τηρούνται αυστηρά για κάθε νέα αίτηση χορήγησης άδειας κυκλοφορίας- οποιαδήποτε απόκλιση πρέπει να αιτιολογείται.
Τα δεδομένα παρουσιάζονται σύμφωνα με τη μορφή του κοινού τεχνικού εγγράφου (CTD), όπως ορίζεται από τη Διεθνή Διάσκεψη για την Εναρμόνιση των Τεχνικών Απαιτήσεων για την Καταχώριση Φαρμάκων για Ανθρώπινη Χρήση (ICH). Η συμφωνία για τη συγκέντρωση όλων των πληροφοριών ποιότητας, ασφάλειας και αποτελεσματικότητας με αυτή την κοινή μορφή (CTD) έφερε επανάσταση στη διαδικασία κανονιστικής αναθεώρησης και οδήγησε σε εναρμονισμένες ηλεκτρονικές υποβολές που, με τη σειρά τους, επιτρέπουν την εφαρμογή ορθών πρακτικών αναθεώρησης. Στη βιομηχανία, εξαλείφθηκε η ανάγκη αναδιαμόρφωσης των πληροφοριών για την υποβολή τους στις διάφορες ρυθμιστικές αρχές (το ICH περιλαμβάνει τις ρυθμιστικές αρχές και τη φαρμακοβιομηχανία της Ευρώπης, της Ιαπωνίας και των ΗΠΑ για να συζητήσουν τις επιστημονικές και τεχνικές πτυχές της καταχώρισης των φαρμάκων).
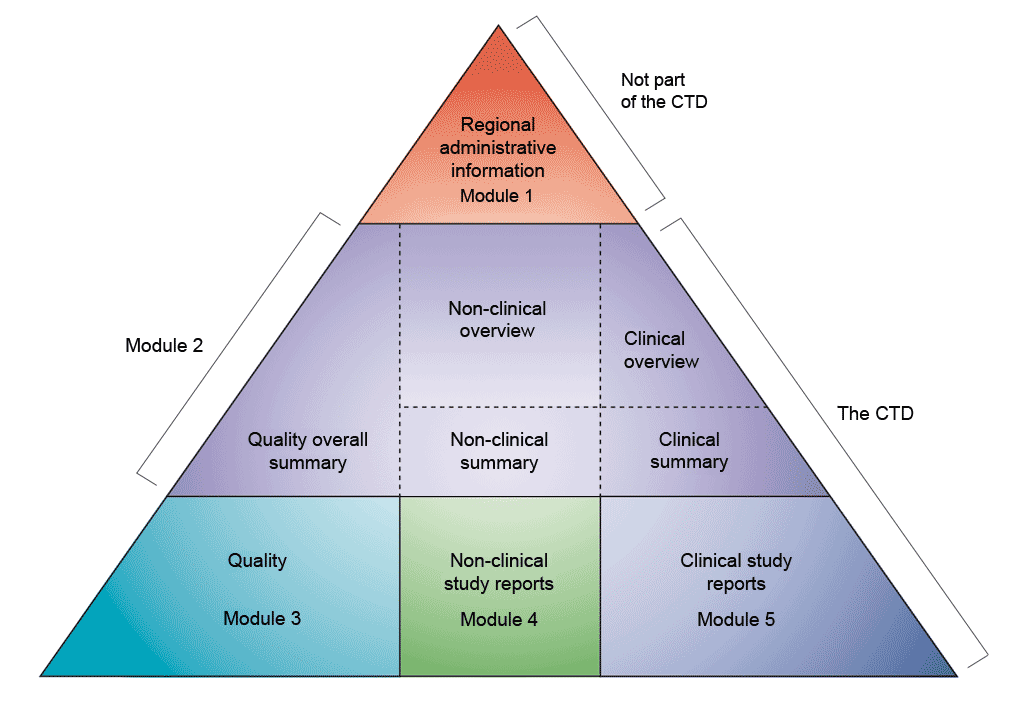
Το κοινό τεχνικό έγγραφο (CTD) είναι οργανωμένο σε πέντε ενότητες (βλέπε σχήμα παραπάνω). Τον Ιούλιο του 2003, το CTD έγινε η υποχρεωτική μορφή για τις νέες αιτήσεις χορήγησης άδειας κυκλοφορίας στην ΕΕ και την Ιαπωνία και η συνιστώμενη μορφή επιλογής για τις αιτήσεις νέων φαρμάκων (NDA) που υποβάλλονται στον Οργανισμό Τροφίμων και Φαρμάκων των ΗΠΑ (FDA).
Σύνοψη
Η φάση της μη κλινικής ανάπτυξης είναι κρίσιμη και πρέπει να προβλέπει πιθανά προβλήματα πριν η ένωση περάσει στη φάση της κλινικής ανάπτυξης.
Η υποβολή μιας υποψήφιας ένωσης σε κλινικές μελέτες απαιτεί:
- Μη κλινική αξιολόγηση της ασφάλειας που πραγματοποιείται υπό συνθήκες ορθής εργαστηριακής πρακτικής (GLP).
- Η παρασκευή να εκτελείται υπό τον κατάλληλο ποιοτικό έλεγχο.
- Τεκμηρίωση δεδομένων και διαδικασιών σύμφωνα με τη μορφή CTD και δημιουργία βάσεων για τη φάση της κλινικής ανάπτυξης.
Υπάρχει μια αυξημένη τάση in silico σχεδιασμού ιδιοτήτων που προσομοιάζουν με φάρμακα, καθώς και χρήσης μεθόδων βιοπληροφορικής για μοντελοποίηση και πρόβλεψη.
Οι κατευθυντήριες γραμμές για τη μη κλινική ανάπτυξη αποτελούν αντικείμενο συνεχούς εναρμόνισης μεταξύ των κύριων ρυθμιστικών αρχών (Ευρώπη, ΗΠΑ και Ιαπωνία) με έμφαση στην ασφάλεια και την ποιότητα: Το ICH εκδίδει τακτικά λεπτομερείς οδηγίες για τη φαρμακοβιομηχανία, παρόμοιες με αυτές που δημοσιεύονται από τους ευρωπαϊκούς (EMA) και αμερικανικούς (FDA) οργανισμούς.
Περαιτέρω πόροι
- Ευρωπαϊκός Οργανισμός Φαρμάκων (2007α). Κατευθυντήρια γραμμή σχετικά με τις στρατηγικές για τον εντοπισμό και τον μετριασμό των κινδύνων για τις πρώτες δοκιμές σε ανθρώπους με ερευνητικά φάρμακα. Λονδίνο: Ευρωπαϊκός Οργανισμός Φαρμάκων. Ανακτήθηκε στις 25 Ιουνίου, 2015, από τη διεύθυνση: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf
- Ευρωπαϊκός Οργανισμός Φαρμάκων (2007β). Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal Ανακτήθηκε στις 25 Ιουνίου, 2015, από τη διεύθυνση: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002989.pdf
- Ευρωπαϊκός Οργανισμός Φαρμάκων (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. Λονδίνο: Ευρωπαϊκός Οργανισμός Φαρμάκων. Ανακτήθηκε στις 25 Ιουνίου, 2015, από τη διεύθυνση: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
- Ευρωπαϊκός Οργανισμός Φαρμάκων (2011). Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. Λονδίνο: Ευρωπαϊκός Οργανισμός Φαρμάκων. Ανακτήθηκε στις 25 Ιουνίου, 2015, από τη διεύθυνση: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
Παραπομπές:
- Η εικόνα αναπαράγεται από το ICH (2015). M4: Το κοινό τεχνικό έγγραφο. Ανακτήθηκε στις 11 Ιουλίου, 2021, από τη διεύθυνση: https://www.ich.org/page/ctd
- Ευρωπαϊκός Οργανισμός Φαρμάκων (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. Λονδίνο: Ευρωπαϊκός Οργανισμός Φαρμάκων. Ανακτήθηκε στις 25 Ιουνίου, 2015, από τη διεύθυνση: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
A2-2.01.1-1.2