Last update: 18 juillet 2023


Principes globaux pour l’implication des patients tout au long du processus de recherche et de développement des médicaments
L’Académie européenne des patients (EUPATI) est un projet paneuropéen d’Initiative Médicaments Innovants (IMI) composé de 33 organisations avec des partenaires d’organisations de patients, des universités, des organisations à but non lucratif et des entreprises pharmaceutiques. Dans toute l’EUPATI, le terme « patient » fait référence à tous les groupes d’âges quelles que soient les conditions. EUPATI ne se concentre pas sur des problèmes ou des traitements propres à des maladies, mais sur le processus de développement des médicaments en général. Les informations propres à une indication ou les interventions médicales spécifiques ou spécifiques à l’âge n’entrent pas dans le champ d’action de l’EUPATI et relèvent des professionnels de santé et des organisations de patients. Pour plus d’informations, consultez eupati.eu/.
La grande majorité des experts impliqués dans le développement et l’évaluation de médicaments sont des scientifiques travaillant dans les secteurs privé et public. Il est de plus en plus nécessaire de faire appel à l’expérience et aux connaissances des patients afin de comprendre ce qu’il en est de vivre avec une condition spécifique, la manière dont les soins sont administrés et l’utilisation des médicaments au quotidien. Cette intervention contribue à améliorer la recherche, le développement et l’évaluation de nouveaux médicaments efficaces.
Une interaction structurée entre patients de tous les groupes d’âges et atteints de diverses conditions, ainsi qu’avec leurs représentants et autres parties prenantes, est nécessaire et permet un échange d’informations et un dialogue constructif aux niveaux national et européen lorsque les points de vue d’utilisateurs de médicaments peuvent et doivent être pris en considération. Il est important de prendre en compte le fait que les systèmes de santé ainsi que les pratiques et la législation peuvent différer.
Nous recommandons une coopération et un partenariat étroits entre les diverses parties prenantes, dont les organisations de professionnels de santé, les organisations de recherche de contrat, les organisations de patients et de consommateurs*, le milieu universitaire, les sociétés scientifiques et universitaires, les autorités réglementaires et les organismes d’évaluation des technologies de la santé (ETS) ainsi que le secteur pharmaceutique. L’expérience démontre à ce jour que l’implication de patients a entraîné un accroissement de la transparence, de la confiance et du respect mutuel entre eux et les autres parties prenantes.
Il est reconnu que la contribution des patients à la recherche, au développement et à l’évaluation de médicaments enrichit la qualité des preuves et des avis disponibles.[1]
Des codes de pratique existants pour l’implication des patients avec diverses parties prenantes ne couvrent pas globalement l’ensemble de la portée de la recherche et du développement (R&D). Les documents de recommandations d’EUPATI visent à prendre en charge l’intégration de l’implication des patients tout au long du processus de recherche et de développement des médicaments.
Ces documents de recommandations ne visent pas à être normatifs et ne fourniront pas de conseils détaillés.
EUPATI a développé ces documents de recommandations pour toutes les parties prenantes cherchant à interagir avec des patients dans la recherche et le développement des médicaments (R&D). Les utilisateurs peuvent s’écarter de ces recommandations du fait d’une législation nationale, de circonstances spécifiques ou des besoins uniques de chaque interaction. Ces recommandations doivent être adaptées pour des exigences individuelles s’appuyant sur le meilleur jugement professionnel.
Il y a quatre documents de recommandations distincts abordant l’implication des patients dans les domaines suivants :
- R&D de médicaments dirigée par les professionnels du domaine pharmaceutique
- Comités d’éthique
- Autorités réglementaires
- Évaluation des technologies de santé (ETS)
Chaque recommandation suggère des domaines offrant actuellement des opportunités d’implication des patients. Ces recommandations doivent être régulièrement analysées et revues pour refléter l’évolution.
Ces recommandations couvrent l’implication des patients dans le domaine réglementaire et s’appuient sur le document éprouvé « Framework for interaction between the European Medicines Agency and patients and consumers and their organisations » (cadre révisé relatif aux interactions entre l’Agence européenne des médicaments et les patients, les consommateurs et les organisations les représentant).
Les valeurs suivantes sont reconnues dans les recommandations et mises en œuvre via l’adoption des pratiques de travail suggérées (section 7). Ces valeurs sont les suivantes :
| Pertinence | Les patients ont des connaissances, des points de vue et des expériences qui sont uniques et contribuent de manière significative à des aspects essentiels d’activités de réglementation. |
| Équité | Les patients disposent des mêmes droits que les autres parties prenantes en matière de contribution aux activités de réglementation et ils ont accès à des connaissances et à des expériences qui permettent une implication efficace. |
| Équité | L’implication des patients dans les activités de réglementation contribue à l’équité en cherchant à comprendre les divers besoins de patients présentant des problèmes de santé spécifiques, en fonction des exigences strictes des directives et de la législation réglementaire. |
| Légitimité | Pour les patients affectés par les décisions réglementaires, l’implication des patients facilite leur participation à des activités réglementaires en contribuant à la transparence, la responsabilité et la crédibilité du processus de prise de décision. |
| Renforcement des capacités | Les processus d’implication des patients gèrent les barrières concernant l’implication de patients dans les activités réglementaires et le renforcement de la capacité pour que les patients et les autorités réglementaires collaborent. |
Toutes les recommandations développées par la suite doivent se conformer à la législation nationale couvrant les interactions comme indiqué dans les quatre documents de recommandations d’EUPATI.
Clause d’exclusion de responsabilité
EUPATI a développé ces recommandations pour toutes les parties prenantes cherchant à interagir avec des patients dans la recherche et le développement des médicaments (R&D) tout au long du cycle de vie de R&D des médicaments.
Ces documents de recommandations ne visent pas à être normatifs et ne fourniront pas de conseils détaillés. Ces recommandations devraient être utilisées du fait de circonstances spécifiques, d’une législation nationale ou de besoins uniques de chaque interaction. Ces recommandations doivent être adaptées pour des exigences individuelles s’appuyant sur le meilleur jugement professionnel.
Si ces recommandations offrent des conseils sur des conditions légales, elles ne sont pas proposées comme interprétation légale définitive et ne remplacent pas un conseil juridique formel. Si un conseil formel est requis, les parties prenantes impliquées doivent consulter leur service juridique respectif le cas échéant, ou demander un conseil juridique auprès de sources compétentes.
EUPATI ne sera en aucun cas responsable de tout résultat, de quelque nature que ce soit, provenant de l’utilisation de ces recommandations.
Le projet EUPATI a reçu le soutien du programme IMI2 JU (Innovative Medicines Initiative Joint Undertaking) dans le cadre de l’accord de subvention n°115334, dont les ressources se composent d’une contribution financière du septième programme-cadre de l’Union Européenne (FP7/2007-2013) et d’entreprises membres de l’EFPIA.
Champ d’application
Ces recommandations européennes couvrent l’interaction entre les patients et les autorités de réglementation des médicaments concernant des médicaments à usage humain. Le terme « Patients » peut faire référence à des patients individuels ou à leurs soignants ou encore à des représentants d’organisations de patients offrant une expertise pertinente (section 4). Les autorités de réglementation incluent à la fois des autorités compétentes nationales (autorités réglementaires nationales) et l’Agence européenne des médicaments (EMA). Les organisations de patients sont sans but lucratif et s’intéressent aux soins des patients, ces derniers représentant une majorité des membres dans les organes directeurs.
Les recommandations insistent sur l’implication et excluent la collection scientifique de points de vue de patients (c’est-à-dire la recherche quantitative et qualitative systématique sur l’impact psychosocial de maladies et de traitements). La figure 1 indique dans quels domaines les patients peuvent actuellement être impliqués tout au long du cycle de vie de la R&D des médicaments. Cela ne vise cependant pas à limiter l’implication et ces opportunités peuvent changer et se développer avec le temps.
Définition du terme « patient »
Le terme « patient » est souvent utilisé de manière générale et imprécise, ne reflétant pas les différents types d'entrées et l'expérience requise des patients, des défenseurs des patients et des organisations de patients dans différents processus de collaboration.
Afin de clarifier la terminologie pour de potentiels rôles d'interactions patients présentés dans ces documents de recommandations et d'autres rédigés par EUPATI, nous utilisons le terme « patient » qui couvre les définitions suivantes :
- Les « patients individuels » sont des individus ayant une expérience personnelle de vie avec une maladie. Ils peuvent avoir ou non des connaissances techniques en matière de R&D ou de processus de réglementation, mais leur rôle principal vise à apporter leur contribution avec leur expérience subjective de la maladie et du traitement.
- Les « soignants » sont des personnes prenant en charge des patients individuels, tels que les membres de la famille ainsi que les aidants rémunérés ou volontaires.
- Les « défenseurs des patients » sont des personnes s'appuyant sur la connaissance et l'expérience du soutien d'une plus large population de patients vivant avec une maladie spécifique. Ils peuvent être affiliés, ou non, à une organisation.
- Les « représentants d'organisations de patients » sont des personnes qui sont mandatées pour représenter et exprimer les points de vue collectifs d'une organisation de patients sur un domaine thérapeutique ou une question spécifique.
- Les « patients experts », en plus d'une expertise spécifique à la maladie, ont une connaissance technique dans la R&D et/ou les affaires réglementaires via la formation ou l'expérience, par exemple des membres d'EUPATI qui ont été formés par EUPATI sur l'éventail complet de la R&D des médicaments.
Du fait que l'intervention de patients individuels sera subjective et ouverte à des critiques, il peut y avoir des réserves quant à leur implication dans des activités en collaboration avec des parties prenantes. Néanmoins, EUPATI, conformément aux autorités réglementaires, insuffle la valeur d'équité en n'excluant pas l'implication d'individus. Le choix de la représentation de patient la plus adéquate concernant le type de patient en fonction de l'activité (voir section 7) doit être laissé à la discrétion de la ou des organisations ayant pris l'initiative de l'interaction. Lorsqu'un patient individuel sera impliqué, il est recommandé que l'organisation de patients pertinente, le cas échéant, soit informée et/ou consultée, pour apporter un soutien et/ou un conseil.
Le type de participation et de mission de la personne impliquée doit être convenu dans tout processus collaboratif avant l'engagement.
Logique de la recommandation
L'étendue de l'implication du patient dans les questions réglementaires varie considérablement entre les pays et les régions en Europe.
L'EMA a interagi avec ses parties prenantes depuis sa création en 1995. Ces relations avec les parties prenantes ont évolué avec le temps, en outre le type et le degré d'interaction varie selon le groupe de parties prenantes concerné et le type d'activité d'EMA. Le Comité de direction de l'EMA et certains comités scientifiques incluent des patients et des consommateurs en tant que membres.
L'avantage de l'implication de la partie prenante expérimenté par l'EMA a entraîné la mise en œuvre par plusieurs organismes réglementaires nationaux d'une structure pour l'implication de patients à un niveau national également. La plupart des régulateurs nationaux s'appuient sur les expériences de l'EMA. L'implication de patients auprès de l'EMA est déterminée par la législation européenne [2]. L'EMA, son comité de direction et ses divers comités scientifiques sont responsables du développement de la relation entre l'EMA et ses parties prenantes.
La législation européenne définit les éléments suivants :
- Interaction directe entre l'EMA et les organisations de patients et de consommateurs, via le Groupe de travail avec les organisations de patients et de consommateurs (PCWP).
- L'infrastructure pour fournir des informations claires et utiles à ces organisations.
- Des formes d'interaction spécifiques, par exemple adhésion des patients au comité de direction de l'EMA, le Comité des médicaments orphelins (COMP), le Comité des médicaments pédiatriques (PDCO), le Comité des médicaments de thérapie innovante (CAT), procédures d'assistance protocole/conseil scientifique avec le Scientific Advice Working Party (SAWP) et le Comité pour l’évaluation des risques en matière de pharmacovigilance (PRAC).
- En outre, l'EMA a mis en place des méthodes pour collecter des données de patients via une consultation directe.
L'expérience acquise à ce jour démontre que la participation des patients à des activités d'EMA a entraîné une transparence accrue et une confiance dans les processus réglementaires ainsi qu'un respect mutuel entre régulateurs et la communauté de patients et de consommateurs. L'expérience confirme l'importance pour EMA de continuer à soutenir et à faciliter la contribution des patients à son travail.
Des dispositions légales similaires peuvent manquer au niveau national. En l'absence de dispositions légales, les autorités compétentes nationales développent leurs infrastructures sur l'expérience d'EMA ou développent leur propre infrastructure. Les éléments clés à prendre en considération pour une telle infrastructure incluent les suivants :
- Définition du rôle des patients dans l'interaction
- Inclusion de propositions sur l'implication de patients dans des processus institutionnels spécifiques
- Développement d'un programme de formation
- Examen d'un concept pour indemnisation d'experts, s'appliquant à toutes les parties prenantes
- Évaluation continue de l'interaction en vue de nouvelles améliorations et développement entre agences avec des patients pour établir et normaliser des méthodes et des pratiques.
Toute infrastructure doit être revue régulièrement.
Objectifs de l'implication de patients dans la réglementation des médicaments
La rationalisation des interactions avec les patients et la concentration sur des domaines dans lesquels un avantage mutuel peut être anticipé sont deux principes sous-jacents à prendre en considération lors de la mise en place d'une infrastructure.
L'objectif doit être de développer davantage la transparence et la confiance avec des communautés de patients via leur engagement actif (participation-consultation-information). Afin d'atteindre ce but, des objectifs spécifiques doivent être atteints, tels que les suivants :
- Soutien du régulateur pour l'accès à des expériences concrètes de maladies et leur gestion et pour l'obtention d'informations sur l'utilisation actuelle de médicaments. Cela contribuera à comprendre la valeur, telle qu'elle est perçue par les patients, de la preuve scientifique fournie pendant le processus d'évaluation aux fins de la prise de décision avantage/risque.
- Garantie que les patients et leurs organisations de représentants sont écoutés, consultés et impliqués dans le développement de stratégies et de plans.
- Amélioration de la compréhension des organisations de patients de la mission et du rôle du régulateur dans le contexte du développement, de l'évaluation, de l'autorisation, de la surveillance et de la fourniture d'informations sur des médicaments.
- Optimisation des outils de communication (par ex. contenu et fourniture) afin de faciliter et d'encourager la cascade d'informations pour les parties liées aux organisations de patients (pour atteindre des patients individuels) en vue de soutenir leur rôle dans l'utilisation sûre et rationnelle de médicaments.
- Facilitation de la participation de patients à l'évaluation bénéfice/risque et activités connexes afin de capturer les préférences et les valeurs de patients et d'obtenir des informations sur l'utilisation actuelle de médicaments et leur environnement thérapeutique, tout au long du cycle de vie du développement des médicaments, depuis le début jusqu'à l'évaluation et la surveillance post-commercialisation.
Pour atteindre ces objectifs, il faudra une collaboration étroite entre les autorités de réglementation, les ministères nationaux de la santé et autres parties prenantes importantes, ainsi qu'une participation active des patients, des professionnels de santé et de leurs organisations de représentants et une bonne interaction de leur part.
Propositions de pratiques de travail (adaptées de l'infrastructure d'interaction d'EMA)
En se basant sur l'expérience de l'EMA au niveau européen, les patients peuvent prendre part aux activités de l'autorité de réglementation en occupant les rôles suivants :
- Membres (et suppléants) de certains des groupes de travail ou comités (scientifiques) d'autorités de réglementation et, dans le cas de l'EMA, du conseil de gestion de l'EMA (anciennement nommé par les institutions de l'UE).
- Experts individuels.
- Représentants d'une organisation spécifique, à consulter et qui participent aux discussions pour exprimer les points de vue de l'organisation sur une question spécifique.
- Occasionnellement, observateurs de certains aspects du travail de l'EMA ou de l'autorité de réglementation.
Les autorités de réglementation devraient établir des critères d'éligibilité.
Lorsque des patients participent à des activités de régulateurs en tant qu'individus et non en tant que représentants de leur organisation, ils doivent déclarer tout intérêt et se conformer au code de conduite du régulateur comme tous les autres experts. En outre, les organisations impliquées auprès du régulateur doivent être entièrement transparentes concernant leurs activités et sources de financement.
Afin d'atteindre les objectifs identifiés sous la section 4, les six éléments suivants devraient être considérés comme critiques :
- Un réseau d'organisations de patients (potentiellement en collaboration avec d'autres autorités de réglementation)Le réseau d'organisations de patients permet au régulateur de développer des interactions cohérentes et ciblées avec un large groupe d'organisations offrant une gamme diversifiée d'expertise et d'intérêts. Des critères de sélection doivent s'appliquer. Ces critères doivent garantir que le régulateur établit un contact avec les organisations les plus adaptées représentant des patients de manière transparente. Dans un réseau, les critères doivent être harmonisés.
- Un forum d'échange avec des organisations de patients établi avec l'autorité de réglementationIl s'agit d'une plateforme pour le dialogue et l'échange avec des organisations de patients sur des questions pertinentes concernant les médicaments à usage humain et, le cas échéant, des appareils médicaux. Par ce biais, le régulateur informera et obtiendra des retours et une contribution de la part de patients sur diverses initiatives de régulateur. Il inclut une représentation équilibrée des différents types de patients ainsi que d'organisations représentant des populations spéciales et vulnérables qui ne sont pas bien représentées dans le développement des médicaments, telles que les personnes âgées et les femmes. Il devrait fournir un forum permettant de mieux identifier les écarts et les priorités sur l'ensemble de l'interaction.
- Un groupe de patients individuels agissant en tant qu'experts dans leur maladie et son traitement pour faciliter l'implication des patients dans les informations et l'évaluation des médicamentsLa création du groupe d'experts permettra au régulateur d'identifier rapidement et efficacement des patients qui peuvent être impliqués dans des activités liées au produit, la révision d'informations du produit et de documents de communication.
- Interaction particulièrement dans le domaine de la communication Cela va fournir une contribution importante pour soutenir les structures existantes pour la diffusion de l'information au public. En outre, la collaboration dans ce domaine va promouvoir la fourniture d'informations validées et à jour sur les avantages et les risques des médicaments et contribuer à la préparation et à la diffusion de messages clairs sur l'utilisation sûre et rationnelle de médicaments visant à atteindre le public. Tous les documents d'information remis aux patients doivent être revus par des représentants des patients afin d'améliorer la lisibilité et l'adéquation du langage et du contenu.
- Un programme d'actions pour le développement de la capacité, insistant sur la formation et la sensibilisation concernant le système de réglementation Pour que leur contribution soit importante, les patients doivent comprendre la mission du régulateur, ainsi que le rôle attendu du patient dans le processus d'évaluation. Un programme de formation doit être disponible. Certaines organisations de patients, ou autres projets de collaboration, ont développé leurs propres documents de formation afin de permettre aux patients de jouer un rôle reconnu de défenseurs des intérêts.
- Support financier Un support financier doit être fourni aux patients contribuant aux activités des régulateurs. Cela représenterait une reconnaissance du travail qu'ils fournissent tout en promouvant leur indépendance. Les patients devraient être reconnus en tant qu'experts et traités selon les mêmes normes que tous les autres experts, également en termes d'indemnisation. Parfois, des patients peuvent avoir besoin d'aide supplémentaire pour s'assurer d'être capables de participer.
Définition de l'interaction
Avant chaque interaction, un accord mutuel doit être établi sur les éléments suivants (le cas échéant) :
- L'objectif de l'activité impliquant des patients et/ou des secteurs d'intérêt commun pour établir une interaction structurée convenue, en fournissant à toutes les parties la protection nécessaire concernant l'indépendance, le secret, la confidentialité et les attentes.
- Le type d'intervention et d'obligation de la personne impliquée.
- Les outils et les méthodes d'interaction, par ex. fréquence de rencontres, règles de base, résolution de conflit, indemnisation, évaluation.
- La méthode d'interaction (rencontres, discussions téléphoniques, etc.) doit être discutée et convenue mutuellement, la commodité pour les patients/organisations de patients étant la priorité. Si l'interaction requiert des rencontres en personne, ou le développement et la fourniture d'événements, ils doivent respecter les codes de conduite en termes de lieu/d'emplacement adéquat et du niveau d'hospitalité fourni.
- Lorsque des événements sont organisés, la capacité à se présenter de tout public de patients visé doit être prise en considération en entreprenant des mesures adéquates pour permettre l'accessibilité, une aide pour le déplacement et l'accès à l'événement.
- Organisation partenaire patient/patient souhaité pour favoriser des relations de travail sur le long terme, avec l'indépendance assurée.
- Profil du type de patient(s) ou de représentant(s) de patients devant être impliqué ainsi que le nombre.
- Mode d'utilisation prévu des résultats de l'activité.
- Manière dont le ou les patients impliqués seront informés des résultats, et moment.
- Termes et conditions contractuels incluant le consentement et la confidentialité, ainsi que l'accord sur l'interaction elle-même (type de rencontre, fréquence, indemnisation).
- Autres éléments en fonction de la spécificité de l'activité.
Identification patient/interaction
Il existe de nombreuses manières d'identifier des patients devant être impliqués dans une interaction. Les voies d'accès principales sont les suivantes :
- Organisations de patients existantes
- EUPATI ou projet similaire
- Publicité autour d'opportunités de participation des patients
- Appel à collaboration ouvert
- Relations existantes avec des prestataires de soins de santé, des hôpitaux et des chercheurs et d'autres agences
- Demandes non sollicitées soumises au préalable par des parties intéressées
- Groupes/Comités consultatifs existants (par ex. Patients and Consumers Working Party à l'EMA, Think Tank EFPIA)
- Agences tierces
Critères d'éligibilité
Associations de patients
Pour accroître la transparence de l'implication des patients, les agences et organisations de patients devraient prévoir de révéler publiquement leurs activités de collaboration sur une base annuelle. Des noms de patients individuels peuvent être révélés lorsque la personne fait partie d'un conseil consultatif générique, mais dans d'autres cas les noms ne doivent pas être communiqués.
Les organisations de patients doivent s'engager à participer activement à l'interaction avec une autorité de réglementation.
Les organisations doivent être établies dans un État membre de l'Union européenne (UE) ou de l'Espace économique européen (EEE) et elles doivent remplir les critères suivants :
| Légitimité : | l'organisation doit avoir des statuts enregistrés dans un des États membres de l'UE/EEE. S'il s'agit d'une organisation internationale non enregistrée dans un État membre de l'UE/EEE, d'autres informations doivent être fournies, apportant la preuve des activités et de l'intérêt de l'UE. |
| Mission/objectifs : | l'organisation ou le patient expert individuel doit avoir une mission/des objectifs clairement définis et accepter qu'ils soient publiés sur le site web des autorités de réglementation. |
| Activités : | l'organisation doit avoir, dans le cadre de ses activités, un intérêt spécifique pour les produits médicaux (et, le cas échéant, les appareils médicaux) devant être documenté (par ex. via un rapport publié sur le site web de l'organisation ou de personnes à titre individuel). |
| Représentation : | l'organisation doit être représentative de patients dans l'ensemble de l'UE/l'EEE ou au niveau national pertinent. Des organisations déjà enregistrées au niveau de la communauté, par ex. dans l'EU Health Forum, le Conseil de l'Europe, sont considéré comme des représentants adéquats des patients ou pour une implication dans des activités de réglementation des médicaments.
En cas de manque d'associations européennes pour un domaine thérapeutique ou une maladie spécifique, l'implication d'organisations nationales peut être considérée, même si la préférence sera accordée aux associations d'envergure européenne. L'éligibilité d'organisations internationales peut également être prise en considération du fait qu'elles jouissent d'une représentation et d'un intérêt européen, y compris le ou les bureaux basés dans l'UE/l'EEE. |
| Structure : | l'organisation doit jouir d'organes directeurs qui sont élus par leurs membres qui doivent être des patients, leurs soignants ou leurs représentants élus. |
| Responsabilité et modalités de consultation : | les déclarations et opinions de l'organisation doivent refléter les avis et points de vue de ses membres et des procédures de consultation adéquates auprès de ces membres doivent être en place. En particulier, l'organisation doit s'assurer que le flux adéquat d'informations est en place pour permettre un dialogue bilatéral : depuis et vers ses membres. |
| Transparence : | l'organisation doit révéler à l'autorité de réglementation ses sources de financement, à la fois publiques et privées, en fournissant le nom des entités et leur contribution financière individuelle, en termes absolus et en termes de pourcentage global du budget de l'organisation. Toute relation avec un promoteur d'entreprise doit être claire et transparente. Ces informations doivent être communiquées à l'agence chaque année.
Dans le cas d'organisations de coordination, la liste des associations membres doit être mise à la disposition de l'agence. L'organisation doit publier sur le site web de l'organisation les statuts enregistrés, ainsi que des informations financières dont ses sources de financement, tant publique que privée, et des informations sur ses activités. L'organisation doit suivre un code de conduite/une stratégie régulant sa relation avec les promoteurs, et son indépendance à leur égard. L'autorité de réglementation va évaluer les informations financières en fonction d'une réglementation prédéfinie transparente. |
Compensation
Il faut reconnaître que globalement les patients impliqués dans des activités le font volontairement, soit à titre individuel, soit en tant que membre d'une organisation. Il faut donc prendre en considération les éléments suivants :
- Indemniser le total du temps investi plus les dépenses.
- Toute indemnisation proposée doit être juste et appropriée en fonction du type d'implication. Idéalement, les coûts de trajet doivent être payés directement par le partenaire en charge de l'organisation au lieu d'être remboursés.
- Couvrir les coûts encourus par les organisations de patients au moment de l'identification ou de la prise en charge de patients pour l'implication dans des activités (à savoir, groupes de soutien par les pairs, formation et préparation).
- Aider à l'organisation de la logistique de la participation des patients, y compris les déplacements et/ou les logements.
L'indemnisation inclut également des avantages indirects en nature (par exemple, l'organisation de patients fournit des services gratuitement) ou tous autres avantages non financiers en nature fournis au patient/à l'organisation de patients (comme des sessions de formation, la configuration de sites web).
Accord écrit
Au minimum, un accord écrit doit définir clairement les éléments suivants : une description de l'activité et de ses objectifs, la nature de l'interaction pendant l'activité, le consentement (le cas échéant), la publication, la confidentialité, l'indemnisation, la confidentialité des données, la conformité, la déclaration de conflit d'intérêts, les délais. L'interaction ne pourra se poursuivre que sur la base d'un accord écrit qui établit au minimum les éléments fondamentaux de la collaboration (par ex., règles d'engagement, conformité, propriété intellectuelle, versements financiers). Il convient de veiller à ce que les accords écrits soient clairs et ne limitent pas le partage de connaissance adéquat.
Mise en application et surveillance
Une structure d'implication du patient peut être introduite étape par étape et/ou à la suite d'une phase pilote, le cas échéant. Après la mise en application complète, lorsque des patients sont impliqués dans des questions générales et spécifiques à un produit, et qu'il y a un groupe établi d'organisations et de patients en tant qu'experts individuels, ainsi que pour des forums d'interaction, un rapport annuel public sur des interactions devrait être préparé, y compris les éléments suivants :
- analyse d'indicateurs (devant être définis pour le type d'interaction) évaluant l'utilité des interactions
- commentaires reçus de patients et de leurs organisations de représentants via des études ciblées
- commentaires reçus de l'autorité de réglementation elle-même
- présentation des activités dans lesquelles des organisations et des patients en tant qu'experts individuels ont été impliqués
- suggestion d'avancée, y compris une stratégie pour de futures interactions de patients, recommandée
Annexe 1 – Feuille de route de l'implication des patients dans des processus de réglementation – exemple EMA
-
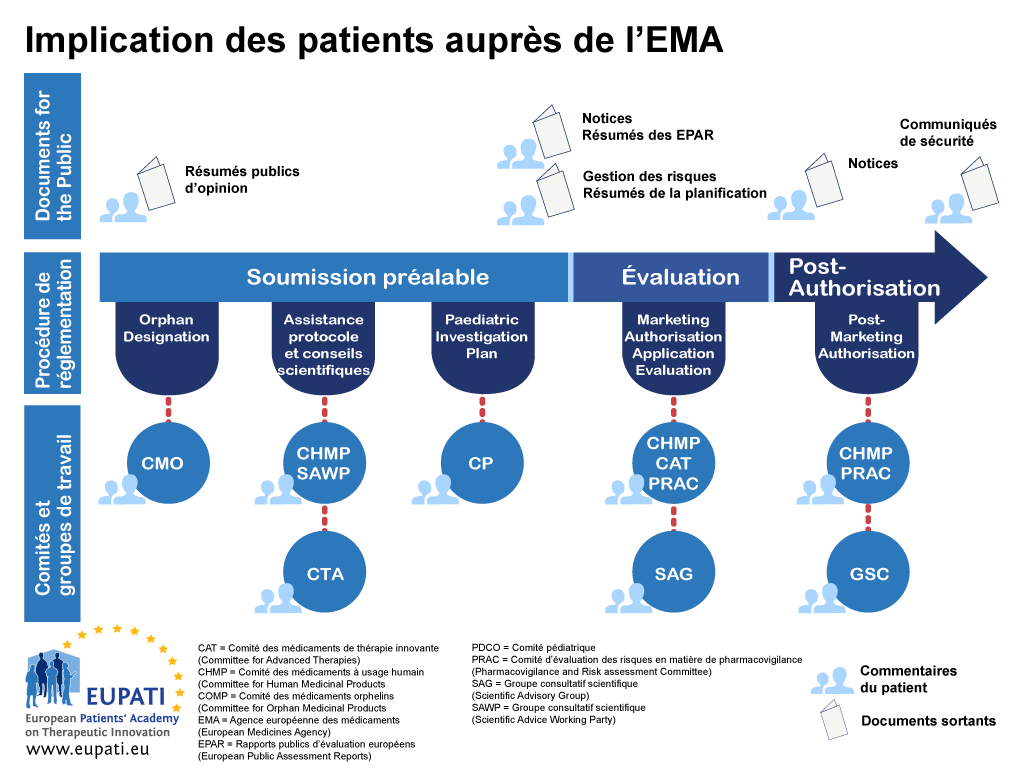
- Les patients peuvent s’engager de différentes façons auprès de l’EMA tout au long de la procédure réglementaire.
Annexe 2 – Codes de pratique revus
Un certain nombre de codes reconnus pourraient fournir une base importante pour ce document de recommandations.
- Le protocole ECAB (description et procédures de fonctionnement de l'ECAB (European Community Advisory Board, groupe de travail scientifique à l'EATG, établi en 1997))
- Les obligations, les objectifs et les règles de procédures pour le groupe de travail des comités scientifiques sur l'usage humain de l'Agence européenne des médicaments avec les organisations de patients et de consommateurs (PCWP) (30 mai 2013)
- Les comptes rendus des réunions du groupe de travail des comités scientifiques sur l'usage humain de l'EMA avec des organisations de patients et de consommateurs (PCWP) avec toutes les organisations admissibles (31 janvier 2014)
- Article de réflexion du 10 décembre 2009 de l'EMA sur la poursuite de l'implication des patients et des consommateurs dans les activités de l'agence
- Dépliant de l'EMA sur le travail avec les patients et les consommateurs (mise à jour : 22/4/2015)
- Cadre d'interaction de l'EMA (révision : 16 octobre 2014)
- Recommandations de la réunion de l'ECAB qui s'est tenue à Bergen, Norvège, en 1997ECAB EATG, « The impatient Patient - From Anger to Activism » (« Le patient impatient - De la colère à l’activisme »)Un examen systématique de l'histoire, des modèles de travail, de l'importance et des perspectives du Conseil consultatif de la Communauté européenne
- Programme du représentant des patients de la FDA
- FDA Patient-Focused Drug Development; The Voice of the Patient (Développement de médicaments focalisés sur le patient; la voix des patients) : une série de rapports issue de l'initiative de développement de médicaments destinée aux patients de la FDA
- FDA Patient-Focused Drug Development (Développement de médicaments focalisés sur le patient) : Enhancing Benefit-Risk Assessment in Regulatory Decision-Making (Augmenter l’évaluation de la balance bénéfice-risque dans le processus de décision réglementaire)
- Déclaration d'Helsinki WMA - Principes éthiques de la recherche médicale impliquant des sujets humains Retrieved 13 July, 2021, from https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/
Références
- Adapted from the EMA framework. European Medicines Agency (2014) EMA/637573/2014. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2009/12/WC500018013.pdf. Last Accessed 21.11.2016.
- Regulation (EC) No 726/2004 http://ec.europa.eu/health/files/eudralex/vol-1/reg_2004_726/reg_2004_726_en.pdf. Last Accessed 21.11.2016.
*Les consommateurs sont reconnus en tant que parties prenantes dans le dialogue de soins de santé. La portée d'EUPATI se concentre sur les patients plus que sur les consommateurs, ce qui se reflète dans les documents de recommandations et le matériel pédagogique.