

Published and available for citation: Haerry D, Landgraf C, Warner K, Hunter A, Klingmann I, May M and See W (2018) EUPATI and Patients in Medicines Research and Development: Guidance for Patient Involvement in Regulatory Processes. Front. Med. 5:230. doi:10.3389/fmed.2018.00230
Overarching principles for patient involvement throughout the medicines research and development process
The European Patients’ Academy (EUPATI) is a pan-European Innovative Medicines Initiative (IMI) project of 33 organisations with partners from patient organisations, universities, not-for-profit organisations, and pharmaceutical companies. Throughout EUPATI the term ‘patient’ references all age groups across conditions. EUPATI does not focus on disease-specific issues or therapies, but on process of medicines development in general. Indication-specific information, age-specific or specific medicine interventions are beyond the scope of EUPATI and are the remit of health professionals as well as patient organisations. To find out more visit eupati.eu/.
The great majority of experts involved in the development and evaluation of medicines are scientists working both in the private and public sector. There is an increasing need to draw on patient knowledge and experience in order to understand what it is like to live with a specific condition, how care is administered and the day-to-day use of medicines. This input helps to improve discovery, development, and evaluation of new effective medicines.
Structured interaction between patients of all age groups and across conditions, their representatives and other stakeholders is necessary and allows the exchange of information and constructive dialogue at national and European level where the views from users of medicines can and should be considered. It is important to take into account that healthcare systems as well as practices and legislation might differ.
We recommend close cooperation and partnership between the various stakeholders including healthcare professionals’ organisations, contract research organisations, patients’ and consumers’ organisations*, academia, scientific and academic societies, regulatory authorities and health technology assessment (HTA) bodies and the pharmaceutical industry. Experience to date demonstrates that the involvement of patients has resulted in increased transparency, trust and mutual respect between them and other stakeholders.
It is acknowledged that the patients’ contribution to the discovery, development and evaluation of medicines enriches the quality of the evidence and opinion available.[1]
Existing codes of practice for patient involvement with various stakeholders do not comprehensively cover the full scope of research and development (R&D). The EUPATI guidance documents aim to support the integration of patient involvement across the entire process of medicines research and development.
These guidance documents are not intended to be prescriptive and will not give detailed step-by-step advice.
EUPATI has developed these guidance documents for all stakeholders aiming to interact with patients on medicines research and development (R&D). Users may deviate from this guidance according to specific circumstances, national legislation or the unique needs of each interaction. This guidance should be adapted for individual requirements using best professional judgment.
There are four separate guidance documents covering patient involvement in:
- Pharmaceutical industry-led medicines R&D
- Ethics committees
- Regulatory authorities
- Health technology assessment (HTA).
Each guidance suggests areas where at present there are opportunities for patient involvement. This guidance should be periodically reviewed and revised to reflect evolution.
This guidance covers patient involvement in the regulatory field and draws on the mature “Framework for interaction between the European Medicines Agency and patients and consumers and their organisations”.
The following values are recognised in the guidance, and worked towards through the adoption of the suggested working practices (section 7). The values are:
| Relevance | Patients have knowledge, perspectives and experiences that are unique and contribute significantly to essential aspects of regulatory activities. |
| Fairness | Patients have the same rights to contribute to the regulatory activities as other stakeholders and have access to knowledge and experiences that enable effective engagement. |
| Equity | Patient involvement in regulatory activities contributes to equity by seeking to understand the diverse needs of patients with particular health issues, balanced against the strict requirements of regulatory legislation and guidelines. |
| Legitimacy | Patient involvement facilitates those affected by regulatory decisions to participate in regulatory activities; contributing to the transparency, accountability and credibility of the decision-making process. |
| Capacity building | Patient involvement processes address barriers to involving patients in regulatory activities and build capacity for patients and regulatory authorities to work together. |
All subsequently developed guidance should be aligned with existing national legislation covering interactions as stated in the four EUPATI guidance documents.
Disclaimer
EUPATI has developed this guidance for all stakeholders aiming to interact with patients on medicines research and development (R&D) throughout the medicines R&D lifecycle.
These guidance documents are not intended to be prescriptive and will not give detailed step-by-step advice. This guidance should be used according to specific circumstances, national legislation or the unique needs of each interaction. This guidance should be adapted for individual requirements using best professional judgment.
Where this guidance offers advice on legal issues, it is not offered as a definitive legal interpretation and is not a substitute for formal legal advice. If formal advice is required, involved stakeholders should consult their respective legal department if available, or seek legal advice from competent sources.
EUPATI will in no event be responsible for any outcomes of any nature resulting from the use of this guidance.
The EUPATI project received support from the Innovative Medicines Initiative Joint Undertaking under grant agreement n° 115334, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and EFPIA companies.
Scope
This European guidance covers the interaction between patients and medicines regulatory authorities in relation to medicines for human use. “Patients” can be individual patients or their carers, or representatives from patient organisations with relevant expertise (section 4). Regulatory authorities include both National Competent Authorities (national regulatory authorities) and the European Medicines Agency (EMA). Patients’ organisations are not-for-profit organisations that have an interest in patient care, and where patients represent a majority of members in governing bodies.
The guidance focuses on involvement, and excludes the scientific collection of patient perspectives (i.e. quantitative and qualitative systematic research on the psychosocial impact of diseases and treatments). Figure 1 indicates where patients can be involved currently throughout the medicines R&D lifecycle; however this is not meant to limit involvement, and opportunities may change and increase over time.
-
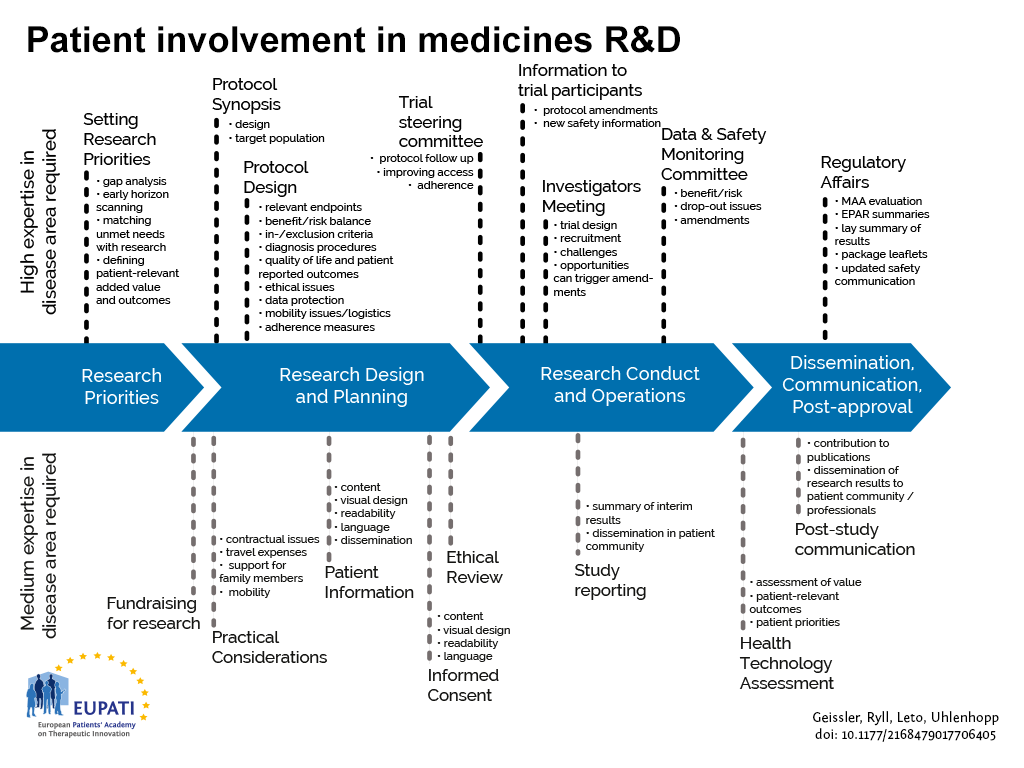
- Patients can be involved across the process of medicines R&D. This diagram created by Geissler, Ryll, Leto, and Uhlenhopp identifies some existing areas in which patients are involved in the process. It distinguishes between the level of expertise in a disease area that is required and the different areas where involvement can take place.
Defining “patient”
The term “patient” is often used as a general, imprecise term that does not reflect the different types of input and experience required from patients, patient advocates and patient organisations in different collaborative processes.
In order to clarify terminology for potential roles of patient interaction presented in this and the other EUPATI guidance documents, we use the term “patient” which covers the following definitions:
- “Individual Patients” are persons with personal experience of living with a disease. They may or may not have technical knowledge in R&D or regulatory processes, but their main role is to contribute with their subjective disease and treatment experience.
- “Carers” are persons supporting individual patients such as family members as well as paid or volunteer helpers.
- “Patient Advocates” are persons who have the insight and experience in supporting a larger population of patients living with a specific disease. They may or may not be affiliated with an organisation.
- “Patient Organisation Representatives” are persons who are mandated to represent and express the collective views of a patient organisation on a specific issue or disease area.
- “Patient Experts”, in addition to disease-specific expertise, have the technical knowledge in R&D and/or regulatory affairs through training or experience, for example EUPATI Fellows who have been trained by EUPATI on the full spectrum of medicines R&D.
There may be reservations about involving individual patients in collaborative activities with stakeholders on grounds that their input will be subjective and open to criticism. However, EUPATI, in line with regulatory authorities, instils the value of equity by not excluding the involvement of individuals. It should be left to the discretion of the organisation/s initiating the interaction to choose the most adequate patient representation in terms of which type of patient for which activity (see section 7). Where an individual patient will be engaged it is suggested that the relevant patient organisation, where one exists, be informed and/or consulted to provide support and/or advice.
The type of input and mandate of the involved person should be agreed in any collaborative process prior to engagement.
Rationale for the guidance
The extent of patient involvement in regulatory issues varies considerably between countries and regions in Europe.
The EMA has interacted with its stakeholders since its creation in 1995. These stakeholder relations have evolved over time and the type and degree of interaction varies depending upon the stakeholder group concerned and the type of EMA activity. The EMA Management Board and certain scientific committees include patients and consumers as members.
The benefit of stakeholder involvement experienced by the EMA has resulted in several national regulatory bodies implementing a framework for involvement of patients at national level too. Most national regulators draw on the EMA experiences.The involvement of patients with the EMA is determined by European legislation [2]. EMA, its Management Board and its various scientific committees are responsible for developing the relationship between the EMA and its stakeholders.
The European legislation defines:
- Direct interaction between the EMA and patients’ and consumers’ organisations, through the Patients’ and Consumers’ Working Party (PCWP),
- The framework for providing clear and useful information to these organisations.
- Specific forms of interaction, e.g. patients’ membership in the EMA Management Board, the Committee for Orphan Medicinal Products (COMP), the Paediatric Committee (PDCO), the Committee for Advanced Therapies (CAT), Scientific Advice/Protocol Assistance procedures with the Scientific Advice Working Party (SAWP) and the Pharmacovigilance and Risk Assessment Committee (PRAC).
- In addition, the EMA has put in place methods to collect patients’ input through direct consultation.
The experience acquired to date demonstrates that the participation of patients in EMA activities has resulted in increased transparency and trust in regulatory processes and mutual respect between regulators and the community of patients and consumers. The experience confirms the importance for EMA to continue supporting and facilitating patient contribution to its work.
Similar legislative provisions may be lacking at the national level. In the absence of legal provisions, National Competent Authorities are developing their frameworks on EMA experience or are developing a framework on their own. Key elements to consider for such a framework include:
- Define the role of patients in the interaction
- Include proposals on involving patients in specific institutional processes
- Develop a training programme
- Consider a concept for expert compensation, applying to all stakeholders
- Continuously evaluate the interaction for further improvements and collaborate among agencies with patients to establish and standardise methods and practices.
Any framework needs to be reviewed on a regular basis.
Objectives of patient involvement in medicines regulation
Streamlining the interactions with patients, and focusing on areas where mutual benefit can be anticipated, are two underlining principles to consider when implementing a framework.
The aim should be further building of transparency and trust with patients’ communities through their active engagement (participation-consultation-information). In order to achieve this goal, specific objectives should be met, such as:
- Supporting the regulator to access real-life experiences of diseases and their management and to obtain information on the current use of medicines. This will contribute to understanding the value, as perceived by patients, of the scientific evidence provided during the evaluation process for the purposes of benefit/risk decision-making.
- Ensure that patients and their representative organisations are listened to, consulted and involved in the development of policies and plans;
- Enhance patients’ organisations understanding of the mandate and role of the regulator within the context of the development, evaluation, authorisation, monitoring and provision of information on medicines;
- Optimise communication tools (on content and delivery) to facilitate and encourage the cascade of information to the constituencies of patients’ organisations (e. to reach out to individual patients) with the aim of supporting their role in the safe and rational use of medicines;
- Facilitate participation of patients in benefit/risk evaluation and related activities, to capture patients’ values and preferences and obtain information on the current use of medicines and their therapeutic environment, all along the lifecycle of medicines development, from early development throughout evaluation and post-marketing surveillance.
Achieving these objectives will necessitate close collaboration between the regulatory authorities, national ministries of health, and other relevant stakeholders, as well as an active participation and good interaction with patients, healthcare professionals and their representative organisations.
Suggested working practices (adapted from the EMA framework of interaction)
Based on experience of the EMA at European level, patients can participate in the regulatory authority’s activities as:
- Members (and alternates) of some of the regulatory authority’s (scientific) committees or working groups and, in case of the EMA, of the EMA’s Management Board (formally appointed by the EU Institutions).
- Individual experts.
- Representatives of a specific organisation, to be consulted and participate in discussions to express the views of the organisation on a specific issue.
- Occasionally observers in certain aspects of the EMA’s or regulatory authority’s work.
Regulatory authorities should establish eligibility criteria.
When patients participate in regulators’ activities as individuals and not as representatives of their organisation, they should declare any interests and abide by the regulator’s code of conduct as any other experts. In addition, the organisations involved with the regulator should be fully transparent with regard to their activities and funding sources.
In order to achieve the objectives identified under section 4, the following six elements should be considered as critical:
- A network of patients’ organisations (potentially in collaboration with other regulatory authorities)
The network of patients’ organisations allows the regulator to build up consistent and targeted interactions with a broad group of organisations with a diverse range of expertise and interests. Selection criteria should apply. Such criteria should ensure that the regulator establishes contact with the most suitable organisations representing patients in a transparent manner. Within a network, the criteria should be harmonised. - A forum of exchange with patients’ organisations established within the regulatory authority
This is a platform for dialogue and exchange with patients’ organisations on relevant issues concerning medicines for human use and when relevant medical devices; through it the regulator will inform and will obtain feedback and contribution from patients on various regulator’s initiatives. It includes a balanced representation of the different types of patients as well as organisations representing special and vulnerable populations not well represented in medicines development such as older people and women. It should provide a forum to further identify gaps and priorities in the overall interaction.
- A pool of individual patients acting as experts in their disease and its treatment to facilitate patients’ involvement in medicines evaluation and information
The creation of the pool of experts will enable the regulator to quickly and efficiently identify patients who can be involved in product-related activities, review of product information and communication material.
- Interaction particularly in the field of communication This will provide a valuable contribution to support the existing structures for information dissemination to the public. Furthermore, collaboration in this area will promote the provision of validated and up-to-date information to patients on the benefits and risks of medicines and contribute to the preparation and dissemination of clear messages on the safe and rational use of medicines intended to reach the public. Any information material to patients should be reviewed by patients’ representatives to improve readability and appropriateness of language and content.
- A programme of actions for capacity-building, focusing on training and raising awareness about the regulatory system For their contribution to be meaningful, patients must have an understanding of the regulator’s mandate as well as the patient’s expected role in the evaluation process. A training programme should be available. Some patients’ organisations or other collaborative projects have developed their own training material in order to empower patients to play a recognised advocacy role.
- Financial support Financial support should be provided to patients contributing to the regulator’s activities. This would represent an acknowledgement of the work they do while promoting their independence. Patients should be recognised as experts and treated according to the same standards as any other experts, also with regard to compensation. Sometimes, patients may need additional assistance to ensure they are able to participate.
Defining the interaction
Prior to each interaction, agree mutually on (where applicable):
- The objective of the activity involving patients and/or areas of common interest to establish an agreed structured interaction, providing all parties with necessary protection with regards to independence, privacy, confidentiality and expectations.
- The type of input and mandate of the involved person
- The tools and methods of interaction, e.g., frequency of meetings, ground rules, conflict resolution, compensation, evaluation.
- The method of interaction (meetings, telephone discussions, etc.) should be discussed and mutually agreed, with convenience for patients/patient organisations as the main priority. If the interaction requires in person meetings or the development and delivery of events, these should follow existing codes of conduct, in terms of appropriate venue/location and the level of hospitality provided.
- When events are organised, the ability of any intended patient audience to attend should be considered, with appropriate measures taken to enable accessibility, assisted travel and entry into the event.
- Desired patient and patient partner organisation to foster long-term working relationships, with independence ensured.
- The profile of the type of patient(s) or patient representative/s to be involved and the number.
- How activity outputs will be used
- How and when the patient/s involved will be informed of outcomes
- Contractual terms and conditions including consent and confidentiality as well as agreement on the interaction itself (type of meeting, frequency, compensation).
- Other elements according to the specific activity
Patient identification / interaction
There are many ways to identify patients to be involved in an interaction. The main routes are through:
- existing patient organisations
- EUPATI or similar project
- advertising opportunities for patient participation
- open call
- existing relationships with healthcare providers, hospitals and researchers and other agencies
- unsolicited requests previously made by interested parties
- existing advisory boards / groups (e.g., Patients and Consumers Working Party at the EMA, EFPIA Think Tank)
- Third party agencies
Eligibility criteria
Patient Organisations
To increase transparency of patient involvement agencies and patient organisations should plan to publicly disclose their collaborative activities on an annual basis. Individual patient names can be disclosed when the person is being part of a generic advisory council but in other instances names should not be disclosed.
Patients’ organisations shall be committed to take an active part in the interaction with a regulatory authority.
The organisations shall be established in a Member State of the European Union (EU) or of the European Economic Area (EEA), and shall fulfil the following criteria:
| Legitimacy: | the organisation shall have statutes registered in one of the Member States of the EU/EEA. If it is an international organisation not registered in an EU/EEA Member State, additional information needs to be provided demonstrating EU focus and activities. |
| Mission/objectives: | the organisation or individual patient expert shall have its /his/hers mission/objectives clearly defined and should agree to have it/them published on the regulatory authorities website. |
| Activities: | the organisation shall have, as part of its activities, a specific interest in medicinal products (and when relevant medical devices) which should be documented (e.g. through a report published on the organisation or individual persons website). |
| Representation: | the organisation shall be representative of patients throughout the EU/EEA or at the relevant national level. Organisations already registered at Community level, e.g. in the EU Health Forum, the Council of Europe, are considered to adequately represent patients or for involvement in medicines regulatory activities.
In case of a lack of European associations for a specific disease or treatment area, the involvement of national organisations may be considered, although preference will be given to European wide-associations. International organisations can also be considered for eligibility as long as they have a European focus and representation, including EU/EEA based office(s). |
| Structure: | the organisation should have governing bodies which are elected by their members, who shall be patients, their carers, or their elected representatives. |
| Accountability and consultation modalities: | statements and opinions of the organisation should reflect the views and opinions of its members and adequate consultation procedures with those members should be in place. In particular, the organisation should ensure that the appropriate flow of information is in place to allow dialogue both ways: from and towards its members. |
| Transparency: | the organisation shall disclose to the regulatory authority its sources of funding both public and private by providing the name of the bodies and their individual financial contribution, both in absolute terms and in terms of overall percentage of the organisation budget. Any relationship with corporate sponsorship should be clear and transparent. This information shall be communicated to the Agency on an annual basis.
In the case of umbrella organisations the list of member associations should be made available to the agency. The organisation shall publish on the organisation website the registered statutes, together with financial information including its source of funding both public and private, and information on their activities. The organisation shall follow a code of conduct/policy regulating its relationship with and independence from the sponsors. The regulatory authority, will evaluate the financial information according to a transparent pre-set regulation. |
Compensation
It should be recognised that in many situations patients involved in activities do so voluntarily either as an individual but also when a member of an organisation. Consideration should therefore be given to:
- compensate for their total time invested plus expenses.
- any compensation offered should be fair and appropriate for the type of engagement. Ideally travel costs would be paid directly by the organising partner, rather than being reimbursed.
- cover the costs incurred by patient organisations when identifying or supporting patients for involvement in activities (i.e peer support groups, training and preparation)
- help organise the logistics of patient participation, including travel and/or accommodation.
Compensation also includes indirect benefits in kind (such as a patient organisation providing services free of charge) or any other non-financial benefits in kind provided to the patient/patient organisation (such as training sessions the setting up of web sites).
Written agreement
At a minimum a written agreement should clearly define: a description of the activity and its objectives, the nature of the interaction during the activity, consent (if relevant), release, confidentiality, compensation, data privacy, compliance, declaration of conflict of interest, timelines. Interaction may only proceed on the basis of a written agreement that at a minimum spells out the basic elements of the collaboration (e.g., rules of engagement, compliance, intellectual property, financial payments). Care should be taken so that written agreements are clear and do not limit appropriate knowledge sharing.
Implementation and monitoring
A patient involvement framework can be introduced step-by-step and/or following a pilot phase where appropriate. After full implementation, when patients are involved both in general and product specific issues and there is an established pool of organisations and patients as individual experts as well as for fora for interaction, a public annual report on interactions should be prepared including:
- an analysis of indicators (to be defined for the type of interaction) assessing the usefulness of the interactions
- feedback received from patients and their representative organisations through targeted surveys
- feedback received from the regulatory authority itself
- an overview of the activities where organisations and patients as individual experts have been involved
- a suggested way forward, including a strategy for future patients’ interaction, is recommended to be proposed.
Appendix 1 – Roadmap on Patient Involvement in regulatory processes – example EMA
-
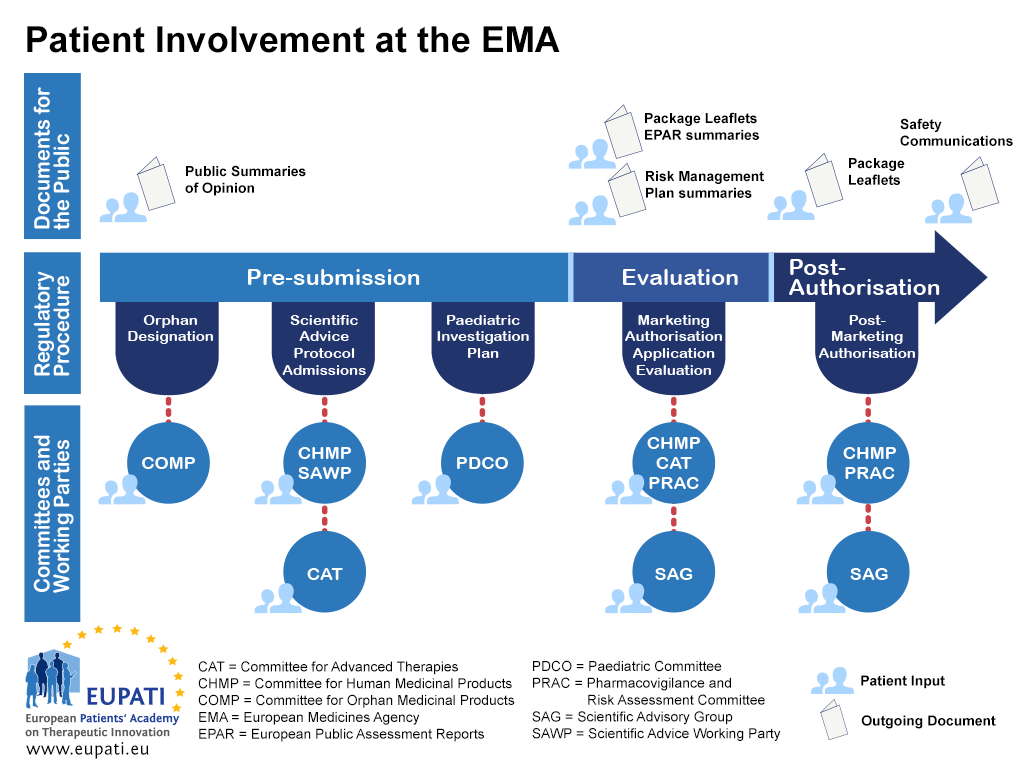
- Patients can be involved at the EMA in a variety of different ways throughout the regulatory procedure.
Appendix 2 – Codes of Practice Reviewed
A number of recognised codes could provide important foundation for this guidance document.
- The ECAB Protocol (description of and working procedures of ECAB (European Community Advisory Board, scientific working group at EATG, established 1997))
- Mandate, objectives and rules of procedure for the European Medicines Agency Human Scientific Committees’ Working Party with Patients' and Consumers' Organisations (PCWP) (30 May 2013)
- Minutes of EMA Human Scientific Committees’ Working Party with Patients’ and Consumers’ Organisations (PCWP) meeting with all eligible organisations (31 January 2014)
- 10 December 2009 EMA Reflection Paper on the Further Involvement of Patients and Consumers in the Agency’s Activities
- EMA leaflet on working with patients and consumers (updated 22/4/2015)
- EMA framework of interaction (revised 16 October 2014)
- Recommendations from ECAB meeting held in Bergen, Norway 1997
EATG ECAB, “The impatient Patient - From Anger to Activism”
A systematic review of the history, working models, relevance and perspectives of the European Community Advisory Board - FDA Patient Representative Program
- FDA Patient-Focused Drug Development; The Voice of the Patient: A Series of Reports from FDA's Patient-Focused Drug Development Initiative
- FDA Patient-Focused Drug Development: Enhancing Benefit-Risk Assessment in Regulatory Decision-Making
- WMA Declaration of Helsinki - Ethical Principles for Medical Research Involving Human Subjects https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ Last Accessed 4 July 2021
References
- Adapted from the EMA framework. European Medicines Agency (2014) EMA/637573/2014. https://www.ema.europa.eu/en/documents/other/revised-framework-interaction-between-european-medicines-agency-patients-consumers-their_en-1.pdf. Last Accessed 4 July 2021
- Regulation (EC) No 726/2004 https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/reg_2004_726/reg_2004_726_en.pdf. Last Accessed 4 July 2021
*Consumers are recognised as stakeholders in the healthcare dialogue. The scope of EUPATI focuses on patients rather than consumers this is reflected in the educational material and guidance documents.