

Biodisponibilité
La biodisponibilité est la fraction (pourcentage) intacte d’un médicament qui atteint la circulation sanguine (circulation systémique).
Dans tous les cas lorsqu’on utilise un médicament, on souhaite que la substance active, ou principe actif, puisse pénétrer dans l’organisme. Toutefois, pour avoir un effet thérapeutique, il ne suffit pas que la substance active pénètre dans l’organisme. Il faut en effet que la dose correcte de substance active soit disponible dans la zone spécifique à traiter. Cette zone spécifique est souvent appelée zone ciblée. En outre, la substance active doit atteindre la zone ciblée en un temps spécifique et y rester pendant une période définie.
Lorsqu’une substance est injectée directement dans la circulation sanguine (injection intraveineuse, IV), la biodisponibilité est de 100 % (voir figure ci-dessous).
-
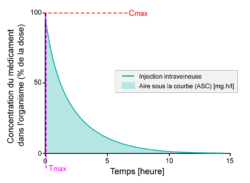
- Pourcentage de principe actif dans le corps ou biodisponibilité après injection directe dans la circulation sanguine, étudié sur une période de 15 heures. L’aire sous la courbe (ASC) est ombrée. Le Tmax est le moment où la concentration du médicament est maximale dans le sang; la Cmax est la concentration maximum du médicament dans le sang.
Après injection, une substance active atteint la zone ciblée après un trajet complexe dans la circulation sanguine. Pour l'évaluation de la biodisponibilité, on mesure la concentration de la substance active dans le sang (circulation systémique) à partir de prélèvements sanguins. Ainsi, la biodisponibilité sera de 100 % directement après l'injection, car la substance active est administrée directement dans le sang. C'est exactement ce que l'on voit sur l'axe des ordonnées (y) de la figure ci-dessus (Biodisponibilité intraveineuse). Ainsi, si on injecte 75 milligrammes (mg) de substance active dans la circulation sanguine, 100 % correspond à 75 mg de substance active.
Lorsque la substance active passe dans la circulation sanguine, une fraction de la substance active sera métabolisée ou excrétée, et de ce fait, la concentration de la substance active dans l'organisme diminuera au fil du temps (voir figure ci-dessus). C'est l'aire sous la courbe (ASC) qui permet d'évaluer le profil de biodisponibilité et de le comparer à celui d'autres médicaments ; l'ASC représente l'exposition totale à une substance active reçue par l'organisme. Le temps nécessaire à l'obtention de la plus forte concentration de substance active dans le sang est le Tmax et la concentration maximale de substance active observée dans la circulation sanguine est la Cmax.
Si la substance active de la figure ci-dessus est administrée par une autre voie, sous forme de comprimé à prise orale par exemple, la biodisponibilité sera inférieure à 100 % (voir figure ci-dessous, Biodisponibilité orale).
-

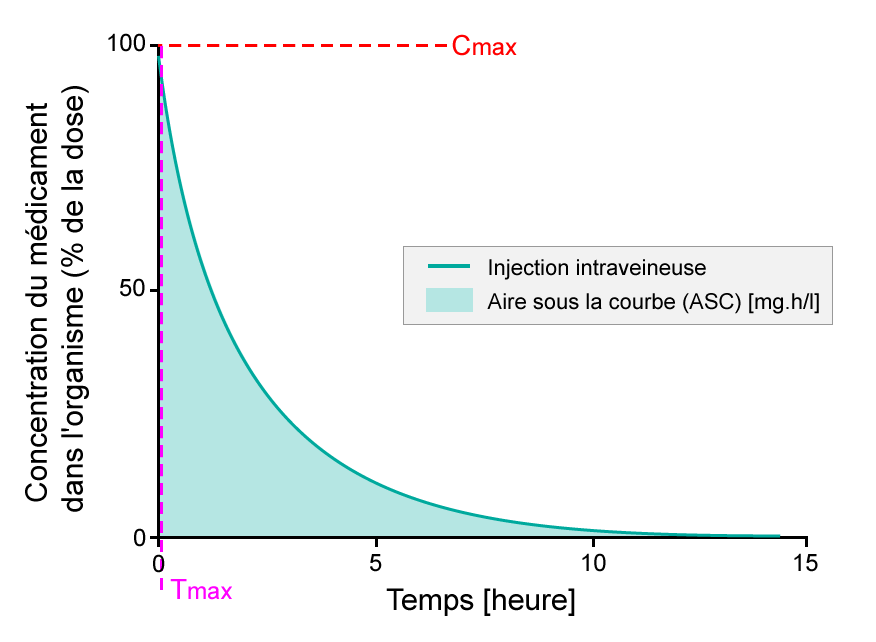
- Pourcentage de principe actif après la prise d’un comprimé, étudié sur une période de 15 heures. L’ASC est ombrée. Le Tmax est le moment où la concentration du médicament est maximale dans la circulation sanguine, tandis que la Cmax est la concentration maximum du médicament dans la circulation sanguine.
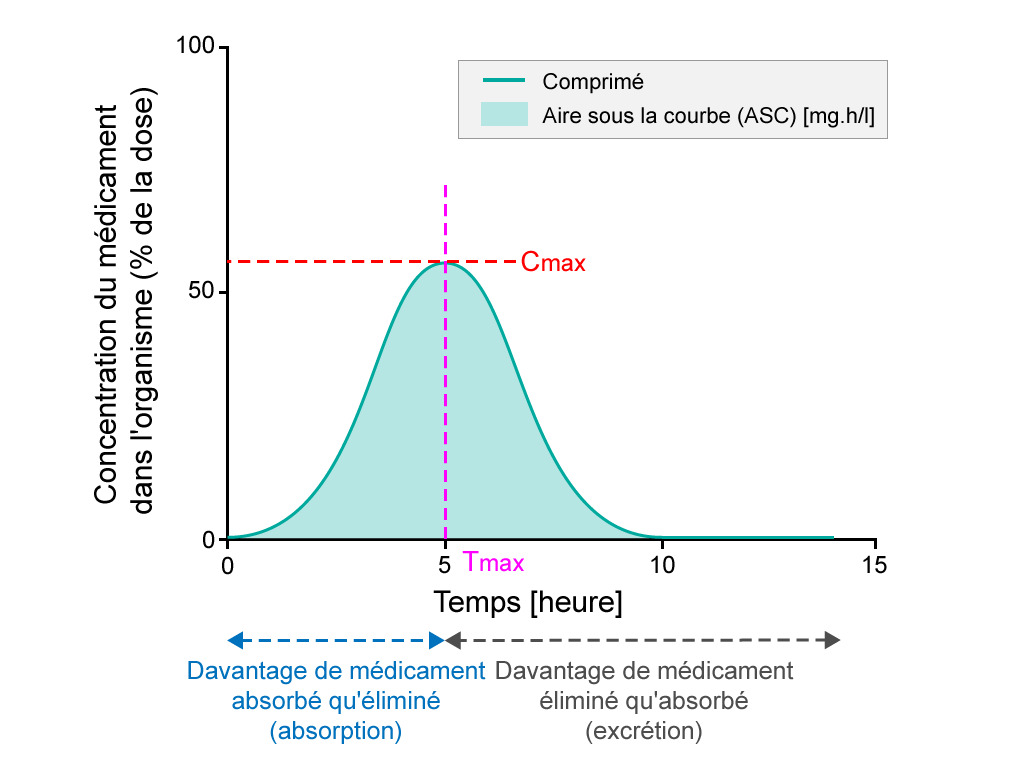
Biodisponibilité orale
Pourcentage de substance active après prise d'un comprimé, étude sur une période de 15 jours. L'ASC est la zone plus foncée. Le Tmax est le temps auquel la plus forte concentration de médicament dans la circulation sanguine est trouvée, tandis que la Cmax est la concentration maximale de médicament observée dans la circulation sanguine.
La biodisponibilité plus faible de la voie orale par rapport à la voie intraveineuse s'explique dans la figure ci-dessous (Biodisponibilité orale et biodisponibilité intraveineuse) :
-
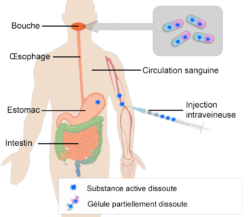
- Diagramme illustrant l’absorption d’une gélule prise par voie orale et une injection directement dans la circulation sanguine (injection intraveineuse). Après avoir atteint l’estomac, la gélule est transportée dans l’intestin grêle ou son absorption se poursuit.
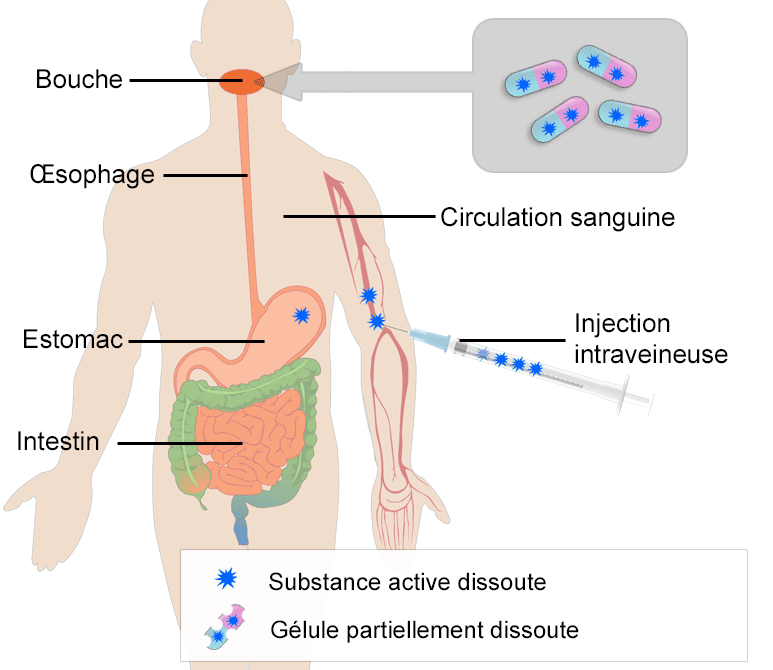
Après la prise d'un comprimé ou d'une gélule, le médicament atteint l'estomac en une ou deux minutes.1 Dans l'estomac, le comprimé ou la gélule se dissout et une partie de la substance active est absorbée dans la circulation sanguine. Les composants sont transportés jusque dans l'intestin grêle, où l'absorption s'achève. L'absorption gastro-intestinale peut être très variable. Une biodisponibilité plus faible peut résulter d'une absorption faible ou nulle dans l'estomac et les intestins ; cette étape est donc un facteur important pouvant affecter la disponibilité.
Lorsque la substance active est absorbée, elle atteint en premier lieu la veine porte et circule jusque dans le foie. La substance active est alors métabolisée pour la première fois par le foie ; c'est l'effet de « premier passage hépatique ». Certaines substances actives sont métabolisées plus largement pendant le premier passage hépatique. La part non métabolisée de la substance active, normalement moins de 100 %, atteint la circulation systémique par la veine hépatique. La quantité qui atteint effectivement la circulation systémique est appelée « biodisponibilité absolue ».
La biodisponibilité absolue compare la biodisponibilité de la substance active dans la circulation systémique après une administration non intraveineuse à la biodisponibilité du même médicament après administration intraveineuse. C'est le pourcentage de substance active absorbé après administration non intraveineuse comparé au même médicament administré par voie intraveineuse.
En bref, pour la biodisponibilité absolue, la norme est l'injection IV.
La biodisponibilité relative mesure la biodisponibilité d'une formulation (A) d'un certain médicament, comparée à celle d'une autre formulation (B) du même médicament, habituellement une autre norme que l'injection IV, ou administrée par une voie différente.
La biodisponibilité est affectée par plusieurs autres facteurs, qui sont spécifiques à chaque individu. Voir des exemples de biodisponibilité dans la fiche technique jointe.
Bioéquivalence
La bioéquivalence est la relation entre deux préparations du même médicament, dans la même présentation, ayant une biodisponibilité similaire.
La biodisponibilité relative permet de comparer non seulement différentes formulations, mais également deux comprimés (ou d'autres médicaments ayant la même formulation) contenant la même substance active, fabriqués par des laboratoires pharmaceutiques différents. Le comprimé du laboratoire A est désigné comme médicament générique du comprimé de référence fabriqué par le laboratoire B (médicament princeps). Pour savoir si le comprimé A est bioéquivalent au comprimé B, on compare le taux de biodisponibilité des deux.2
Ressources complémentaires
- Food and Drug Administation (2002). Guidance for industry: Bioavailability and bioequivalence studies for orally administered drug products – General considerations. Rockville, MD: Food and Drug Administration. Consulté le 23 juin 2015, sur le site http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/UCM154838.pdf
- Wang, H., Li, Q., Reyes, S., Zhang, J., Xie, L., Melendez, V., Hickman, M. and Kozar, M.P. (2013). Formulation and particle size reduction improve bioavailability of poorly water-soluble compounds with antimalarial activity. Malaria Research and Treatment, Consulté le 23 juin 2015, sur le site http://dx.doi.org/10.1155/2013/769234
- Johnson, J.A. (2000). Predictability of the effects of race or ethnicity on pharmacokinetics of drugs. International Journal of Clinical Pharmacology and Therapeutics, 38, 53-60.
Références
- Tatum, R.P., Shi, G., Manka, M.A., Brasseur, J.G., Joehl, R.J. and Kahrilas, P.J. (2000). Bolus transit assessed by an esophageal stress test in postfundoplication dysphagia. Journal of Surgical Research, 91, 56–60.
- MobiSystems, Inc. (2007). Dorland's Medical Dictionary for Health Consumers. [Mobile application software].
Annexes
A2-1.16-V1.2