Last update: 16 июня 2015


Под разработкой лекарственной формы подразумевается процесс переработки активного вещества в готовый к использованию лекарственный препарат, который можно назначать в определенной дозировке по мере необходимости. Разработка лекарственных форм ведется в соответствии с принципами приготовления и составления медицинских препаратов, которые направлены наоптимизацию их усвоения. Это особый раздел фармацевтики, дисциплина (или наука) о создании лекарственных форм.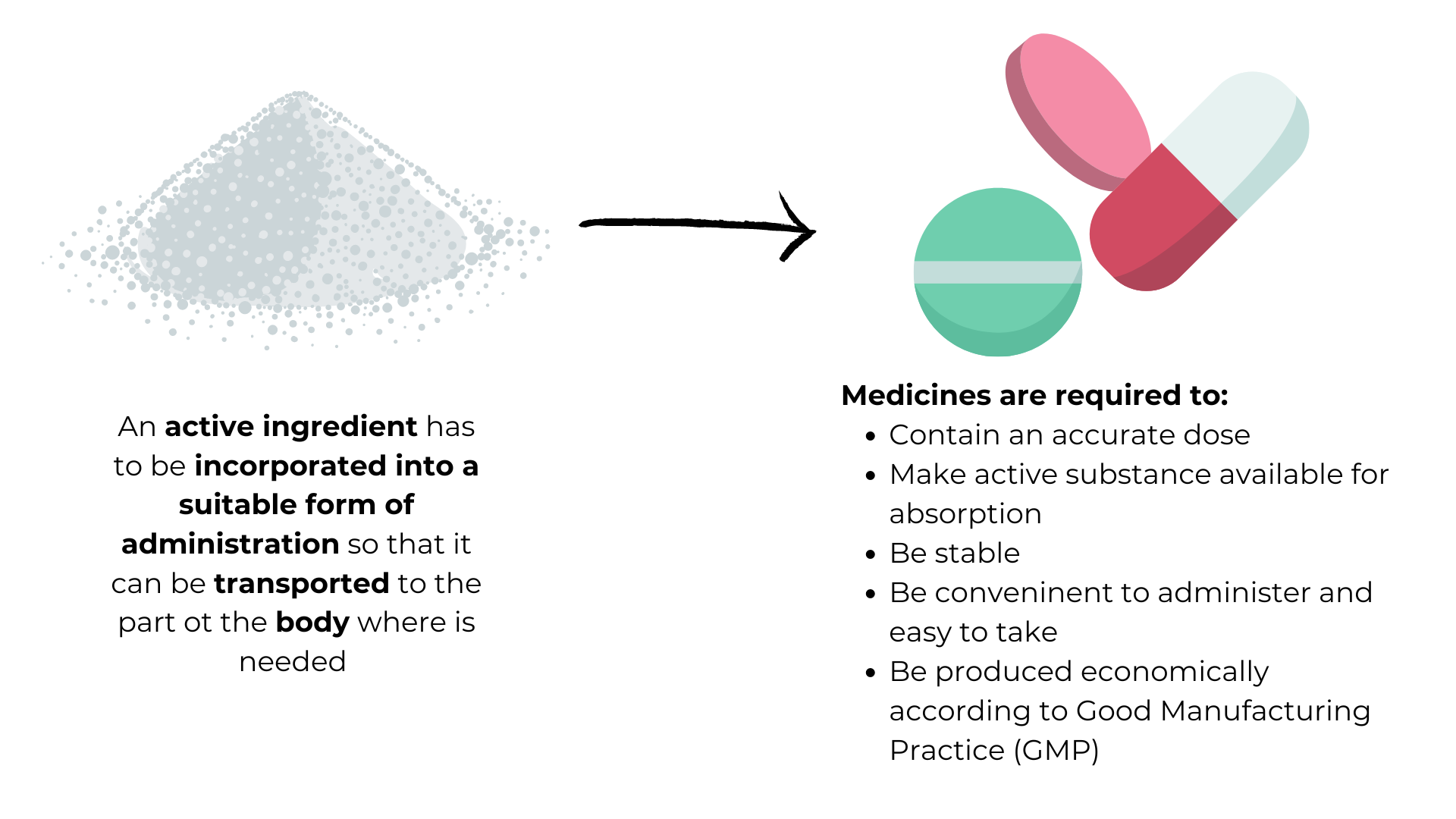
- На рисунке демонстрируется процесс разработки лекарственных форм — внедрения активного ингредиента в готовые к применению медицинские препараты — и приведены требования, предъявляемые к таким медицинским препаратам.
Для разработки лекарственной формы, исследований для оценки безопасности и клинических исследований требуется активное вещество. В результате разработки химического процесса производства лекарственной субстанции производится достаточное количество качественного активного вещества, которое используется для тестирования его безопасности и предоставляется ученым для дальнейшей разработки медицинского препарата.
Медицинский препарат состоит из активного вещества в определенной лекарственной форме (таблетка, мазь, суспензия,раствор), вспомогательных веществ (нейтральных компонентов, таких, как лактоза), и формы упаковки и (или) введения (блистернаяупаковка, бутылка, ингалятор, ампула, заполненный шприц).
Разработка лекарственных форм для доклинических исследований
Доклинические испытания безопасности направлены на
- изучение реакции на вводимые дозы вплоть до максимально допустимых;
- обнаружение потенциальных факторов риска;
- получение объема данных, достаточного для проведения оценки рисков;
- содействие определению дозирования препарата на начальных этапах клинических исследований;
- предварительное определение «маркеров» для мониторинга безопасности препарата в человеческом организме;
- предоставление оснований для целенаправленных специализированных исследований.
Однако проведение указанных исследований не может полностью гарантировать безопасность использования препаратов для человеческого организма или предопределить его реакцию на препарат. При тестировании препарата на безопасность и определяются безопасные границы для получения исследовательских данных на организмах животных и людей. При проведении этого вида испытаний отслеживаются следующие параметры:
- вводимая доза,
- степень и длительность системного воздействия препарата,
- ежедневное системное воздействие,
- воздействие и идентификация метаболитов и
- воздействие на целевые органы.
Испытания на безопасность проводятся как в отношении активного вещества, так и всех сопутствующих веществ, растворителей, продуктов распада, вспомогательных веществ и прочих активных материалов и экстрагирующих компонентов, которые входят в окончательный состав лекарственной формы.
Разработка лекарственных форм для клинических исследований
При разработке подходящей формы введения препарата необходимо учитывать, в частности, такие факторы, как его переносимость пациентом и особенности активного вещества. Нейтральные или вспомогательные вещества могут составлять большую часть объема медицинского препарата, являясь носителем для активного компонента. Например, в таблетках используются кукурузный крахмал или лактоза, а в мазях — водно-масляные эмульсии.
От формы введения препарата также зависит усвоение и доступность активного вещества, а следовательно, и терапевтический эффект препарата. Форма введения определяет, каким образом активное вещество поступает в организм, где и в какой дозировке оно высвобождается и сколько времени необходимо для его усвоения. Кроме того, способ введения препарата должен предусматривать, что пациент сможет безопасно для себя дозировать препарат, и его прием не вызовет у него затруднений.
Специалисты по разработке лекарственных форм следят за тем, чтобы препарат усваивался организмом, а терапевтическая доза достигала необходимого органа. Некоторые активные вещества не подходят для приема внутрь в виде таблетки, в связи с чем специалисты по разработке лекарственных форм постоянно сталкиваются с новыми задачами по соблюдению особых требований по введению препаратов (инъекции в главное яблоко, продукты для ингаляций и таблетки для рассасывания).
Правила организации производства и контроля качества (стандарт GMP)
Производство всех предназначенных для людей медицинских препаратов, в том числе дженериков и биоаналогов, регулируется Правилами организации производства и контроля качества (стандартом GMP).
Стандарт GMP содержит требования относительно производства, тестирования и соблюдения качества препарата с целью обеспечить его безопасный прием для людей. Отдельные законы и нормативно-правовые акты во многих странах требуют от фармацевтических компаний и производителей медицинской техники соблюдения стандарта GMP. Большинство положений Правил GMP содержат требования, согласованные на международном уровне.
Стандарт GMP основан на нескольких фундаментальных принципах:
- Гигиена: На территориях предприятий по производству фармацевтической продукции должны соблюдаться чистота и гигиена.
- Контроль состояния окружающей среды для предотвращения загрязнения медицинской продукции другими составами или твердыми примесями, которые могут сделать медицинский препарат опасным для приема людьми.
- Четко определенные и контролируемые технологии производства:
- Все наиболее значимые процессы подлежат валидации для обеспечения соответствия техническим требованиям и их соблюдения.
- Любые изменения в технологии подлежат проверке.
- Любые изменения, которые оказывают какое-либо влияние на качество медицинского препарата, должны проходить процесс валидации.
- Инструкции и стандарты излагаются четким и ясным языком. Надлежащая практика ведения документации (стандарт GDP)
- Операторы обучаются выполнять производственные процессы и их фиксировать их документально.
- В ходе производства вручную или с помощью инструментовведутся записи, которые демонстрируют, что все этапы, предусмотренные определенными инструкциями и стандартами, действительно соблюдены, и что качество и количество лекарственного препарата соответствует ожидаемому.
- Любые отклонения рассматриваются и фиксируются документально.
- Сведения о производстве (включая сбыт), которые позволяют отследить полную историю производства партии продукции, хранятся в понятной и доступной форме.
- Организация сбыта медицинских препаратов минимизирует все потенциальные риски относительно их качества.
- Существующая система позволяет отозвать любую партию медицинских препаратов из продажи или склада.
- Жалобы на медицинские препараты, находящиеся в продаже, рассматриваются, причины дефектов качества расследуются, в отношении медицинских препаратов с дефектами качества принимаются соответствующие меры, которые направлены в том числе и на предупреждение повторения ситуации.
Дополнительные источники
- Правила организации производства и контроля качества (стандарт GMP) Европейского союза
- Правила организации производства и контроля качества (стандарт GMP) Всемирной организации здравоохранения
A2-2.06-V1.4