Last update: 18 Juli 2023


Allgemeine Grundsätze der Patientenbeteiligung am gesamten Arzneimittelforschungs- und entwicklungsprozess
Die Europäische Patientenakademie (EUPATI) ist ein Projekt der gesamteuropäischen innovativen Arzneimittelinitiative [Innovative Medicines Initiative (IMI)]; sie besteht aus 33 Organisationen mit Partnern in Patientenorganisationen, Universitäten, gemeinnützigen Organisationen und Pharmaunternehmen. EUPATI verwendet den Begriff ‘Patient’ immer für alle Altersgruppen und Krankheiten. EUPATI befasst sich nicht mit krankheitsspezifischen Themen oder Therapien, sondern mit dem Arzneimittelentwicklungsprozess im Allgemeinen. EUPATI befasst sich nicht mit anwendungsspezifischen Informationen, altersspezifischen oder bestimmten Arzneimittelinterventionen; diese Themen fallen in den Aufgabenbereich der Fachkräfte im Gesundheitswesen und der Patientenorganisationen. Sie erfahren mehr darüber auf der folgenden Website eupati.eu/.
Der Großteil der Experten, die an der Entwicklung und Bewertung von Arzneimitteln teilnehmen, sind sowohl im privaten wie auch im öffentlichen Bereich tätig. Es ist zunehmend nötig auf das Wissen und die Erfahrungen von Patienten zurückzugreifen; zu verstehen, was es bedeutet mit einer bestimmten Krankheit zu leben, wie die Betreuung klappt, und über den täglichen Gebrauch von Arzneimitteln zu lernen. Diese Beiträge helfen bei der Entdeckung, Entwicklung und Bewertung neuer, wirksamer Arzneimittel.
Gut organisierte Beziehungen zwischen Patienten aller Altersgruppen und mit allen möglichen Krankheiten, deren Vertretern und anderen Interessengruppen sind notwendig; sie ermöglichen den Informationsaustausch und einen konstruktiven Dialog auf nationaler und europäischer Ebene, und sie berücksichtigen die Erfahrungen der Patienten, bei denen diese Arzneimittel bereits zum Einsatz kamen und kommen. Es ist wichtig nicht zu vergessen, dass Gesundheitssysteme, die medizinische Praxis und Gesetze anderer Ansicht sein können.
Wir empfehlen die enge Zusammenarbeit und Partnerschaft mit verschiedenen Interessengruppen, wie Organisationen für Angehörige der Gesundheitsberufe, Organisationen, die Auftragsforschung betreiben, Patienten- und Konsumentenorganisationen*, akademische Kreise, wissenschaftliche und akademische Gesellschaften, Zulassungsbehörden, Gremien für Gesundheitstechnologiebewertung und die Pharmaindustrie. Die Erfahrungen haben gezeigt, dass Patientenbeteiligung die Transparenz, das Vertrauen und den gegenseitigem Respekt zwischen Patientenorganisationen und anderen Interessengruppen erhöht.
Es ist heute allgemein akzeptiert, dass die Mitwirkung von Patienten an der Entdeckung, Entwicklung und Bewertung von Arzneimitteln dazu beigetragen hat, dass sich die Qualität der Belege und der Bewertung verbessert hat.[1]
Die bestehenden Verhaltensregeln für Patientenbeteiligung an verschiedenen Interessengruppen befassen sich nicht ausführlich mit dem gesamten R&D Bereich. Die EUPATI-Anleitungsdokumente beschäftigen sich damit die Integration der Patientenbeteiligung in allen Prozessen der Arzneimittelforschung- und entwicklung zu fördern.
Diese Anleitungsdokumente sind nicht als verbindlich zu betrachten; es werden keine schrittweisen Anweisungen gegeben.
EUPATI hat diese Anleitungsdokumente für alle Interessengruppen entwickelt, um die Interaktion mit Patienten in der Arzneimittelforschung- und entwicklung (R&D) zu fördern. Diese Anleitung kann an spezifische Umstände, nationale Gesetze oder an eine besondere Aufgabenstellung einer Wechselbeziehung angepasst werden. Diese Anleitung sollte auf individuelle Ansprüche unter Anwendung der besten professionellen Erfahrung abgestimmt werden.
Es gibt vier separate Anleitungsdokumente für Patientenbeteiligung:
- Von der Pharmaindustrie geleitete Arzneimittel-R&D
- Ethik-Kommissionen
- Zulassungsbehörden
- Gesundheitstechnologiebewertung (HTA).
Jede Anleitung enthält Vorschläge für eine mögliche Patientenbeteiligung. Die Anleitungen sollten regelmäßig überprüft und aktualisert werden.
Diese Anleitung umfasst die Patientenbeteiligung im Zulassungsbereich und greift auf den ausgefeilten „Rahmen der Wechselbeziehungen zwischen der europäischen Arzneimittel-Agentur und den Patienten und Konsumenten und der Organisationen zurück”.
Die nachfolgenden Werte werden in der Anleitung respektiert. Durch Annahme der empfohlenen Arbeitsmethoden wird auf deren Verwirklichung hingearbeitet (Abschnitt 7). Die Werte sind wie folgt:
| Relevanz | Patienten haben einzigartiges Wissen, Perspektiven und Erfahrung und leisten wichtige Beträge zu den wesentlichen Aspekten der Zulassungsaktivitäten. |
| Fairness | Patienten haben dieselben Rechte wie andere Interessengruppen Beiträge zu den Zulassungsaktivitäten zu leisten; ihre Erkenntnisse und Erfahrungen ermöglichen es ihnen sich erfolgreich zu beteiligen. |
| Gleichheit | Patientenbeteiligung an den Zulassungsaktivitäten trägt zur Gleichstellung bei, weil versucht wird die unterschiedlichen Bedürfnisse von Patienten mit speziellen Gesundheitsproblemen zu verstehen und mit den strengen Anforderungen der gesetzlichen Regelungen und Leitlinien in Einklang zu bringen. |
| Legitimität | Patientenbeteiligung erleichtert denjenigen die Teilnahme an den Zulassungsaktivitäten, die von den Zulassungsentscheidungen betroffen sind, und trägt zur Transparenz, Rechenschaftspflicht und Glaubwürdigkeit des Entscheidungsprozesses bei. |
| Kapazitätsbildung | Patientenbeteiligungsprozesse befassen sich mit Hindernissen der Patientenbeteiligung an den Zulassungsaktivitäten; sie erweitern die Zusammenarbeit zwischen Patienten und Zulassungsbehörden. |
Wie in allen vier EUPATI Anleitungsdokumenten erwähnt, sollte sich jede zukünftige Anleitung nach den nationalen Gesetzen über Wechselbeziehungen richten.
Haftungsausschluss
EUPATI hat diese Anleitung für alle Interessengruppen entwickelt, um die Beziehungen zwischen Patienten und Arzneimittel-R&D im R&D Stadium von Arzneimitteln zu fördern.
Diese Anleitungsdokumente sind nicht als verbindlich zu betrachten; es werden keine schrittweisen Anweisungen gegeben. Diese Anweisung sollte an spezifische Umstände, nationale Gesetze oder an eine besondere Aufgabenstellung einer Wechselbeziehung angepasst werden. Diese Anleitung sollte auf individuelle Ansprüche unter Anwendung der besten professionellen Erfahrung abgestimmt werden.
Die Empfehlungen in dieser Anleitung bezüglich gesetzlicher Fragen sind nicht als verbindliche rechtliche Interpretationen aufzufassen; sie sind kein Ersatz für eine formelle Rechtsberatung. Sollte formelle Rechtsberatung erforderlich sein, dann sollten sich die jeweiligen Interessengruppen mit ihren Rechtsabteilungen, falls vorhanden, beraten, oder die Rechtsberatung kompetenter Stellen suchen.
In keinem Fall übernimmt EUPATI die Verantwortung für Ergebnisse wie auch immer, die sich aus der Verwendung dieser Anleitung ergeben.
Das EUPATI Projekt erhielt Unterstützung vom Innovative Medicines Initiative Joint Undertaking [gemeinsamer Plan für eine innovative Arzneimittelinitiative] unter Förderungsvertrag Nr. 115334, dessen Ressourcen sich aus finanziellen Beiträgen vom Seventh Framework Programme [7. Rahmenprogramm] der Europäischen Union (FP7/2007-2013) und von EFPIA Firmen zusammensetzen.
Geltungsbereich
Diese europäische Anleitung beschäftigt sich mit der Wechselbeziehung von Patienten und Zulassungsbehörden für Arzneimittel in Bezug auf Arzneimittel für den menschlichen Gebrauch. „Patienten” können Einzelpersonen, deren Betreuer, oder Vertreter von Patientenorganisationen mit relevanter Erfahrung sein (Abschnitt 4). Die Zulassungsbehörden sind die National Competent Authorities (nationalen zuständigen Zulassungsbehörden) und die Europäische Arzneimittel-Agentur [EMA]. Patientenorganisationen sind gemeinnützige Organisationen und haben ein Interesse an Patientenpflege, und dort wo Patienten eine Mehrheit von Mitgliedern in den Führungsgremien haben.
Diese Anleitung konzentriert sich auf die Teilnahme; sie beschäftigt sich nicht mit der wissenschaftlichen Erfassung von Patientenperspektiven (d.h. es werden keine quantitativen und qualitativen systematischen Untersuchungen über die psychosoziale Bedeutung von Krankheiten und Behandlungen durchgeführt). Siehe Abbildung 1, die zeigt, wo sich Patienten derzeit im gesamten R&D Lebenszyklus beteiligen können; damit ist aber nicht gemeint, dass sich die Teilnahme auf diese Bereiche beschränkt; die Teilnahmemöglichkeiten können sich ändern und neue Gelegenheiten können sich eröffnen.
Definition des Begriffs „Patient”
Der Begriff „Patient” wird oft allgemein und ungenau verwendet und berücksichtigt nicht die Unmenge von Beiträgen und Erfahrungen, die von Patienten, Patientenvertretern und den an kollaborativen Prozessen beteiligten Patientenorganisationen gefordert wird.
Um die in dieser und anderen EUPATI Anleitungen beschriebenen Rollen dieser Wechselbeziehungen verständlich zu machen - definieren wir das Wort „Patient” wie folgt:
- „Individuelle Patienten” sind Personen, die persönliche Erfahrung mit einer Krankheit haben. Sie haben vielleicht nur wenig Wissen über R&D oder das Zulassungsverfahren; ihre Hauptaufgabe ist es über ihre Erfahrung mit der Krankheit und der Pflege zu informieren.
- „Betreuer” sind Personen, die individuellen Patienten Beistand leisten, z. B. die Familie und bezahlte oder ehrenamtliche Helfer.
- „Patientenvertreter” sind Personen, die Einblicke in und Erfahrung über spezifische Krankheiten infolge ihrer Arbeit mit Patientengruppen haben. Möglicherweise arbeiten sie mit einer Organisation zusammen.
- „Vertreter einer Patientenorganisation” sind Personen, deren Aufgabe es ist den Standpunkt einer Patientenorganisation zu einem bestimmten Thema oder einer Krankheit zu vertreten und auszudrücken.
- „Patientenexperten” haben abgesehen von einer krankheits-spezifischen Expertise auch das technische Wissen in R&D und/oder Training und Erfahrung in regulatorischen Angelegenheiten, z. B. die EUPATI Fellows; sie erhielten ein Training über das gesamte Spektrum der Arzneimittelforschungs- und entwicklung (R&D) in von EUPATI veranstalteten Kursen.
Interessengruppen werden vielleicht gegen die Teilnahme einzelner Patienten an kollaborativen Aktivitäten protestieren, weil ihre Beiträge subjektiv sind und kritisch beurteilt werden könnten. Der von EUPATI und den Zulassungsbehörden geförderte Einschluss von Einzelpersonen bedeutet, dass sie als gleichwertig angesehen werden. Es sollte dem Ermessen der Organisation, die diese Wechselbeziehung in die Wege leitet, überlassen bleiben die zweckmäßigste Art der Vertretung für die jeweilige Aktivität zu wählen (siehe Abschnitt 7). Es wird empfohlen, dass im Fall der Teilnahme einzelner Patienten die zuständige Patientenorganisation, falls vorhanden, informiert und/oder zurate gezogen und/oder konsultiert wird.
Vor jeder Beteiligung an kollaborativen Prozessen sollte die Art des Beitrags und das Thema mit der teilnehmenden Person abgestimmt werden.
Warum die Anleitung entwickelt wurde
Der Umfang der Patientenbeteiligung an Zulassungsfragen ist in europäischen Ländern und Regionen sehr unterschiedlich.
Die EMA hat seit ihrer Gründung in 1995 mit Interessengruppen zusammengearbeitet. Diese Beziehungen mit den Interessengruppen haben sich über Jahre hindurch entwickelt, und die Art und Intensität der Wechselbeziehung hängt von der betreffenden Interessengruppe und der Art der EMA Aktivität ab. Der EMA Vorstand und bestimmte wissenschaftlichen Ausschüsse schließen Patienten und Konsumenten als Mitglieder ein.
Der Vorteil der Beteiligung von Interessengruppen hat in der Erfahrung der EMA dazu geführt, dass mehrere nationale Zulassungsbehörden einen Rahmen für die Beteiligung von Patienten auf nationaler Ebene geschaffen haben. Die meisten nationalen Behörden greifen auf die Erfahrungen der EMA zurück. Patientenbeteiligung an der EMA ist durch europäischen Gesetze geregelt [2]. Die EMA, der Vorstand und die verschiedenen wissenschaftlichen Ausschüsse sind für die Entwicklung von Beziehungen zwischen der EMA und den Interessengruppen verantwortlich.
Die europäischen Gesetze definieren:
- Direkte Wechselbeziehungen zwischen der EMA und Patienten und Konsumentenorganisationen, durch die Arbeitsgruppe für Patienten- und Konsumenten(PCWP),
- Ist der Rahmen diesen Organisationen klare und nützliche Informationen zu bieten.
- Spezifische Formen der Wechselbeziehung, z. B. die Mitgliedschaft von Patienten im EMA Vorstand, die Kommission for medizinische Waisenprodukte [Committee for Orphan Medicinal Products (COMP)], die pädiatrische Kommission [Paediatric Committee (PDCO)], die Kommission für fortgeschrittene Therapien [Committee for Advanced Therapies (CAT)], wissenschaftliche Beratung/Verfahren zur Mithilfe an Protokollen mit der wissenschaftlichen Arbeitsgruppe [Scientific Advice Working Party (SAWP)], und die Pharmakovigilanz- und Risikobewertungs-Kommission [Pharmacovigilance and Risk Assessment Committee (PRAC)].
- Darüberhinaus hat die EMA auch Methoden eingeführt, die es ihr erlaubt Patientenbeiträge durch direkte Beratung zu sammeln.
Die bisherigen Erfahrungen haben gezeigt, dass Patientenbeteiligung an EMA Aktivitäten die Transparenz, das Vertrauen in den Zulassungsprozess und den gegenseitigem Respekt zwischen Zulassungsbehörden, der Patientengemeinde und Konsumenten erhöht. Diese Erfahrungen bestätigen, dass es für die EMA wichtig ist die Patientenbeiträge zu ihrer Arbeit weiterhin in unterstützen und zu erleichtern.
Es kann sein, dass es an ähnlichen gesetzlichen Regelungen auf nationaler Ebene mangelt. Wegen des Fehlens gesetzlicher Regelungen, entwickeln die nationalen kompetenten Behörden [National Competent Authorities] selbst einen Rahmen. Zu den Schlüsselelementen, die in einem solchen Rahmen berücksichtigt werden sollten, gehören:
- Definition der Rolle der Patienten in der Wechselbeziehung
- Vorschläge beifügen, wie Patienten an spezifischen institutionellen Prozessen teilnehmen können
- Entwicklung eines Trainingsprogramms
- Ein Konzept für Expertenvergütung erwägen, das auf alle Interessengruppen zutrifft
- Ständig die Wechselbeziehung auf weitere Verbesserungen überprüfen, und innerhalb der Vertretungen mit Patienten zusammenarbeiten, um Methoden und Praktiken festzulegen und zu standardisieren.
Jeder Rahmen muss regelmäßig überprüft werden.
Ziele der Patientenbeteiligung an der Zulassung von Arzneimitteln
Die Wechselbeziehungen mit Patienten sollten vereinfacht werden, und sich auf Gebiete konzentrieren, wo beiderseitiger Nutzen zu erwarten ist; das sind die zwei zugrundeliegenden Prinzipien, die bei der Implementierung des Rahmens beachtet werden sollten.
Das Ziel sollte die Entwicklung von Transparenz und Vertrauen mit den Patienten Communitys durch deren aktive Beteiligung (Teilnahme-Beratung-Information)sein. Um dieses Ziel zu erreichen, sollten spezifische Zielsetzungen erreicht werden, wie z. B:
- Unterstützung der Zulassungsbehörden beim Zugriff auf Erfahrungen aus der Praxis mit Krankheiten, und wie Patienten mit diesen Krankheiten leben, sowie Informationen über die gegenwärtige Verwendung von Arzneimitteln. Das trägt zum Verständnis des Wertes der wissenschaftlichen Beweise, wie er von Patienten empfunden wird, während des Bewertungsprozess bei, zum Zweck der Nutzen/Risiko Entscheidungsfindung.
- Sicherstellen, dass die Patienten und ihre Vertreter-Organisationen an der Entwicklung der Regeln und Pläne angehört, konsultiert und beteiligt werden;
- Das Verständnis der Patientenorganisationen über den Auftrag und die Rolle der Zulassungebehörden im Zusammenhang mit der Entwicklung, Bewertung, Zulassung, Überwachung und Kundmachung von Informationen über Arzneimittel vergrößern.
- Die Kommunikationsmittel optimieren (Inhalt und Verbreitung), um die Flut von Informationen an die Mitglieder der Patientenorganisationen (e. um individuelle Patienten zu kontaktieren) zu vereinfachen und sie anzuregen, mit dem Ziel sie bei der ihrer Aufgabe in der sicheren und rationellen Anwendung von Arzneimitteln zu unterstützen;
- Vereinfachung der Patiententeilnahme an der Nutzen/Risiko Bewertung und ähnlichen Aktivitäten, um die Werte und Wünsche der Patienten zu erfassen, und Informationen über die derzeitige Verwendung von Arzneimitteln und das therapeutische Umfeld, über den ganzen Lebenszyklus der Arzneimittelentwicklung, von der frühen Entwicklung bis zur Sicherheitsüberwachung nach Markteinführung, zu erhalten.
Um diese Ziele zu erreichen, ist eine enge Zusammenarbeit mit den Zulassungsbehörden, den nationalen Gesundheitsministerien und anderen relevanten Interessengruppen notwendig, sowie eine aktive Teilnahme und gute Wechselbeziehung mit Patienten, Angehörigen der Gesundheitsberufe und deren Vertretungsorganisationen.
Empfohlene Arbeitspraktiken (übernommen aus dem EMA Rahmen der Wechselbeziehungen)
Aufgrund der Erfahrung der EMA auf europäischer Ebenen, können Patienten an den Aktivitäten der Zulassungsbehörden wie folgt teilnehmen:
- Mitglieder (und Stellvertreter) einiger (wissenschaftlicher) Kommissionen oder Arbeitsgruppen der Zulassungsbehörde, und im Fall der EMA, des EMA Vorstands (formell von den EU Institutionen ernannt).
- Individuelle Experten.
- Vertreter einer spezifischen Organisation, sollte hinzugezogen werden und an Diskussionen teilnehmen, um den Standpunkt der Organisation zu einem bestimmten Thema auszudrücken.
- Manchmal Beobachter bei bestimmten Aspekten der Arbeit der EMA oder der Zulassungsbehörde.
Die Zulassungsbehörden sollten Eignungskriterien festlegen.
Wenn Patienten an Zulassungsaktivitäten als Einzelpersonen und nicht als Vertreter ihrer Organisation teilnehmen, sollten sie ihre Interessen bekannt geben, und wie alle anderen Experten an den Verhaltenskode der Zulassungsbehörde gebunden sein. Darüberhinaus sollten die beteiligten Organisationen vollkommen transparent sein bezüglich ihrer Aktivitäten und Zuwendungen.
Um die in Abschnitt 4 identifizierten Ziele zu erreichen, sollten die Patientenorganisationen die folgenden 6 Elemente kritisch betrachte:
- Ein Netzwerk der Patientenorganisationen (möglicherweise in Zusammenarbeit mit anderen Zulassungsbehörden)Das Netzwerk der Patientenorganisationen ermöglicht es der Zulassungsbehörde einheitliche und gezielte Wechselbeziehungen mit einer breiten Gruppe von Organisationen mit einem vielfältigen Spektrum an Erfahrung und Interessen zu entwickeln. Auswahlkriterien sollten zutreffen. Diese Kriterien stellen sicher, dass die Zulassungsbehörde Kontakt mit den geeignetsten Patientenorganisationen in transparenter Form aufnimmt. Innerhalb eines Netzwerks sollten die Kriterien harmonisiert werden.
- Ein Austauschforum mit Patientenorganisationen sollte innerhalb der Zulassungsbehörden eingeführt werden.Es handelt sich um eine Plattform für Dialog und Meinungsaustausch mit Patientenorganisationen über relevante Fragen bezüglich Arzneimitteln für den menschlichen Gebrauch und, falls relevant, medizinische Produkte; die Zulassungsbehörde kann auf diese Weise informieren und Feedback und Beiträge von Patienten über verschiedene Zulassungsinitiativen erhalten. Sie umfasst eine ausgewogene Repräsentation der verschiedenen Arten von Patienten wie auch Organisationen, die spezielle und gefährdete Bevölkerungsgruppen, die nicht gut in der Arzneimittelentwicklung repräsentiert sind, wie ältere Menschen und Frauen. Sie sollte ein Forum sein, um weitere Lücken und Prioritäten in der gesamten Wechselbeziehung zu identifizieren.
- Eine Gruppe von individuellen Patienten, die sich als Experten in ihrer Krankheit und Behandlung betätigen, um die Beteiligung von Patienten an der Beurteilung von Arzneimitteln und den Informationen zu ermöglichen.Die Schaffung dieser Expertengruppe ermöglicht es der Zulassungsbehörde Patienten sehr schnell und effizient zu identifizieren, die an produktbezogenen Aktivitäten, der Überprüfung der Produktinformation und der Kommunikationsmaterialien teilnehmen können.
- Eine Wechselbeziehung besonders im Bereich der Kommunikation Das ist ein sehr wertvoller Beitrag bei der Unterstützung bestehender Strukturen zur Verbreitung von Informationen an die Öffentlichkeit. Weiterhin fördert die Zusammenarbeit auf diesem Gebiet die Bereitstellung validierter und neuester Informationen an die Patienten über die Vorteile und Risiken von Arzneimitteln, und trägt zur Erstellung und Verbreitung klarer Mitteilungen über den sicheren und rationalen Gebrauch von Arzneimitteln bei, die für die Öffentlichkeit bestimmt sind. Alle Informationen für die Patienten sollten von Patientenvertretern überprüft werden, um die Verständlichkeit und Angemessenheit der Sprache und des Inhalts zu verbessern.
- Ein Aktionsprogramm zur Kapazitätsbildung mit dem Fokus auf Training und Kenntnisse über das Zulassungssystem Damit die Teilnahme sinnvoll ist, müssen die Patienten über den Auftrag der Zulassungsbehörde und auch über die vom Patienten erwartete Rolle im Bewertungsprozess Bescheid wissen. Ein Trainingsprogramm sollte verfügbar sein. Einige Patientenorganisationen oder andere kollaborative Projekte haben ihr eigenes Trainingsmaterial entwickelt, um Patienten zu ermächtigen eine anerkannte Befürworterrolle zu spielen.
- Finanzielle Unterstützung Die Patienten sollten finanzielle Unterstützung für ihre Arbeit an Zulassungsaktivitäten erhalten. Das wäre eine Anerkennung für ihre Arbeit, während es gleichzeitig ihre Unabhängigkeit fördert. Patienten sollten als Experten anerkannt und nach dem gleichen Standard behandelt werden wie andere Experten, auch hinsichtlich der Vergütung. Manchmal ist es möglich, dass Patienten weitere Hilfe benötigen, um sicherzustellen, dass die teilnehmen können.
Definition einer Wechselbeziehung
Vor jeder Wechselbeziehung sollte man gegenseitig Folgendes vereinbaren (wo zutreffend):
- Es ist das Ziel der Aktivität mit Patienten und/oder gemeinsamen Interessengebieten eine akzeptierte strukturierte Wechselbeziehung aufzubauen, und allen Parteien den notwendigen Schutz bezüglich Unabhängigkeit, Vertraulichkeit und Erwartungen zu geben.
- Die Art der Beiträge und der Auftrag der beteiligten Person
- Die Werkzeuge und Methoden für die Wechselbeziehung, z. B. die Art und Häufigkeit der Treffen, Grundregeln, Vergütung, Bewertung.
- Die Art der Wechselbeziehung (Treffen, Telefongespräche, etc.) sollte diskutiert und gegenseitig vereinbart werden; das Hauptaugenmerk sollte auf den Komfort des Patienten/der Patienten gerichtet sein. Falls die Wechselbeziehung persönliche Treffen oder die Vorbereitung und Durchführung von Veranstaltungen erfordert, sollten die bestehenden Verhaltenskodizes und die jeweiligen Gesetze bezüglich eines angemessenen Veranstaltungsorts/Standorts und des Ausmaßes der Gastfreundschaft befolgt werden.
- Falls Veranstaltungen organisiert werden, sollte darauf geachtet werden, dass die geplante Patientenbeteiligung auch möglich ist; die notwendigen Maßnahmen bezüglich Erreichbarkeit, Reisehilfe und Barrierefreiheit sollten getroffen werden.
- Erwünschter Patient und erwünschte Patientenorganisation zur Förderung langfristiger Beziehungen mit garantierter Unabhängigkeit.
- Das Profil und die Art des(r) beteiligten Patienten oder des(r) Patientenvertreter und ihre Anzahl.
- Wie die Aktivitätsbeiträge verwendet werden
- Wie und wann der/die beteiligte(n) Patient(en) über die Ergebnisse informiert werden
- Vertragsbedingungen einschließlich Einwilligung und Vertraulichkeit wie auch eine Vereinbarung über die Wechselbeziehung selbst (Art des Treffens, Häufigkeit, Vergütung).
- Andere Elemente gemäß der spezifischen Aktivität
Patientenidentifizierung/Wechselbeziehung
Es gibt viele Möglichkeiten Patienten zu identifizieren, um sich an einer Wechselbeziehung zu beteiligen Die wichtigsten Möglichkeiten bieten:
- bestehende Patientenorganisationen
- EUPATI oder ein ähnliches Projekt
- Ausschreibung der Gelegenheit zur Patiententeilnahme
- offene Ausschreibung
- bestehende Beziehungen mit Gesundheitspersonal, Krankenhäusern, Forschern und anderen Vertretungen
- unaufgeforderte Anträge von interessierten Parteien
- bestehende Beratungsgremien/Gruppen (z. B. Patienten und Konsumenten-Arbeitsausschuss bei der EMA, EFPIA Think Tank)
- Vertretungen von Drittparteien
Eignungskriterien
Patientenorganisationen
Um die Transparenz der Patientenbeteiligung zu erhöhen, sollten Vertretungen und Patientenorganisationen planen ihre kollaborativen Aktivitäten jährlich auf ihren Webseiten zu veröffentlichen. Die Namen einzelner Patienten können veröffentlicht werden, wenn der Patient Mitglied eines generischen Beratungsgremiums ist, aber in anderen Fällen sollten sie nicht offengelegt werden.
Patientenorganisationen sollen sich verpflichten eine aktive Rolle in der Wechselbeziehung mit Zulassungsbehörden zu spielen.
Die Organisationen sollen in einem Mitgliedsstaat der Europäischen Union [European Union] (EU) oder des Europäischen Wirtschaftsraums [European Economic Area (EEA)] eingerichtet werden und die folgenden Kriterien erfüllen:
| Legitimität: | Die Organisation soll die Statuten haben, die in einem Mitgliedsstaat der EU/EWG registriert sind. Wenn es sich um eine internationale Organisation handelt, die nicht in einem Mitgliedsstaat der EU/EWG registriert ist, müssen die folgenden Informationen vorgelegt werden, die zeigen, dass der Fokus auf die EU und deren Aktivitäten gerichtet ist. |
| Mission/Zielsetzungen: | Die Organisation oder der individuelle Patientenexperte soll ihre/seine Mission/Zielsetzungen klar definieren und sollte zustimmen, dass sie auf der Webseite der Zulassungsbehörden veröffentlicht wird/werden. |
| Aktivitäten: | Die Organisation soll als Bestandteil ihrer Aktivitäten ein spezielles Interesse an Medizinprodukten (und wo relevant, an medizinischen Geräten) haben; das sollte dokumentiert werden (z. B. in einem Bericht über die Organisation oder auf der Webseite der Einzelperson). |
| Repräsentation: | Die Organisation soll die Patienten in der gesamten EU/EWG oder auf relevanter nationaler Ebene repräsentieren. Organisationen, die bereits auf Community Ebene registriert sind, z. B. im EU Gesundheitsforum [Health Forum], dem Europäischen Rat [Council of Europe], werden als Organisationen eingestuft, die Patienten ausreichend repräsentieren oder an Zulassungsaktivitäten über Arzneimittel teilnehmen können.
Falls es keine europäischen Vereine für eine spezifische Krankheit oder ein Behandlungsgebiet gibt, kann die Beteiligung nationaler Organisationen erwogen werden, obwohl europaweiten Organisationen der Vorrang gegeben wird. Internationale Organisationen können auch als geeignet in Betracht kommen, wenn sie einen Fokus auf Europa und die entsprechende Repräsentation haben, einschließlich Büro/s in der EU/EWG. |
| Struktur: | Die Organisation sollte Leitungsorgane haben, die von den Mitgliedern gewählt werden, welche Patienten, deren Betreuer, oder ihre ausgewählten Vertreter sein sollen. |
| Verantwortlichkeit und Beratungsmodalitäten: | Erklärungen und Meinungen der Organisation sollten die Ansichten und Meinungen ihrer Mitglieder reflektieren, und ausreichende Beratungsverfahren mit diesen Mitgliedern sollten vorhanden sein. Vor allem sollte die Organisation sicherstellen, dass es einen angemessenen Informationsfluss gibt, um den Dialog zu ermöglichen: von und zu ihren Mitgliedern. |
| Transparenz: | Die Organisation sollte die Herkunft ihrer öffentlichen und privaten Finanzmittel der Zulassungsbehörde vorlegen, und die Namen der Gremien und deren individuelle finanzielle Beiträge sowohl in absoluten Zahlen als auch als Gesamtprozentsatz des Budgets der Organisation angeben. Jede Beziehung mit Unternehmenssponsoren sollte klar und transparent sein. Diese Information wird jährlich an die Agentur weitergeleitet.
Im Fall von Dachorganisationen, sollte die Liste der Mitgliedsverbände der Agentur zur Verfügung gestellt werden. Die Organisation soll auf ihrer Webseite die registrierten Statuten veröffentlichen zusammen mit finanziellen Informationen, einschließlich der Herkunft der öffentlichen und privaten Finanzmittel, und Informationen über ihre Aktivitäten. Die Organisation soll eine(n) Verhaltenskodex/Richtlinie befolgen, der/die Beziehungen mit und die Unabhängigkeit von den Sponsoren regelt. Die Zulassungsbehörde wird die Finanzinformation überprüfen gemäß transparenter vorab festgelegter Regeln. |
Vergütung
Es sollte hervorgehoben werden, dass die an den Aktivitäten beteiligten Patienten sich oft ehrenamtlich entweder als Einzelpersonen oder auch als Mitglieder von Organisation betätigen. Es ist daher auch Folgendes zu berücksichtigen:
- Die Teilnehmer sollten für Zeit und Spesen entschädigt werden.
- Die Entschädigung sollte fair sein und sich nach der Art der Tätigkeit richten. Im Idealfall sollten die Reisekosten direkt von der Organisation, die die Beteiligung fördert, gezahlt anstatt rückerstattet werden.
- Die Kosten für die Patientenorganisationen zu übernehmen, wenn diese sich an der Identifizierung und Unterstützung von Patienten bei der Teilnahme an Aktivitäten beteiligen (z. B. Peer-Unterstützungsgruppen, Training und Vorbereitung).
- Logistische Unterstützung für eine Patiententeilnahme, einschließlich Reise- und/oder Hotelkosten, sollte gewährt werden.
Unter Kostenerstattung sind auch indirekte Sachleistungen (z. B. eine Patientenorganisation bietet Leistungen kostenlos an) oder andere nicht-finanzielle Leistungen für Patienten/Patientenorganisationen (z. B. Trainingsveranstaltungen, Einrichtung von Webseiten) zu verstehen.
Schriftliche Vereinbarung
Eine schriftliche Vereinbarung sollte zumindest Folgendes klar definieren: Beschreibung der Aktivität, Ziele, und Art der Wechselbeziehung während der Aktivität, Zustimmung (falls zutreffend), Freigabe, Vertraulichkeit, Vergütung, Datenschutz, Compliance, Erklärung von Interessenkonflikten, Zeitpläne. Die Wechselbeziehung darf nur auf Grundlage einer schriftlichen Vereinbarung stattfinden, und soll zumindest die wesentlichen Voraussetzungen für eine Zusammenarbeit regeln, wie z. B. Einsatzregeln ("rules of engagement"), Compliance, geistiges Eigentum, finanzielle Leistungen. Es sollte darauf geachtet werden, dass die schriftlichen Vereinbarungen eindeutig sind und einen zweckmäßigen Wissensaustausch ("knowledge sharing") nicht beschränken.
Implementierung und Überwachung
Ein Rahmen für Patientenbeteiligung kann stufenweise und/oder gegebenenfalls nach einer Pilotphase eingeführt werden. Nach der vollen Implementierung, wenn Patienten sowohl an allgemeinen wie auch an produkt-spezifischen Fragen beteiligt sind, und der Beweis erbracht wurde, dass eine festgesetzte Gruppe von Organisationen und Patienten als Patientenexperten wie auch Foren für Wechselbeziehungen bestehen, sollte ein öffentlicher jährlicher Bericht über Wechselbeziehungen verfasst werden, einschließlich:
- einer Untersuchung der Indikatoren (definiert für die Art der Wechselbeziehung) unter Beurteilung der Nützlichkeit der Wechselbeziehungen
- Feedback von Patienten und ihrer Vertreter-Organisationen durch gezielte Umfragen
- Feedback von der Zulassungsbehörde selbst
- eines Überblicks über Aktivitäten, an denen Organisationen und Patienten als individuelle Experten beteiligt waren
- Es wird empfohlen einen Weg in die Zukunft vorzuschlagen, einschließlich einer Strategie für eine Wechselbeziehung für zukünftige Patienten.
Appendix 1 – „Roadmap“ für die Patientenbeteiligung an Zulassungsverfahren – Beispiel EMA
-
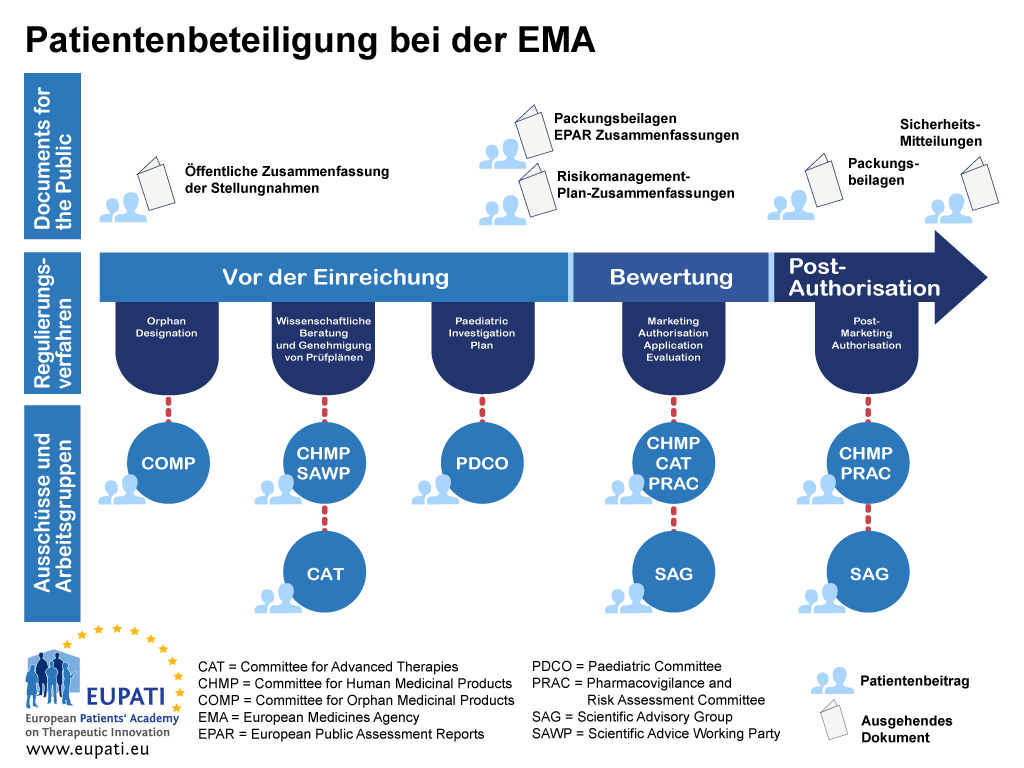
- Patienten können bei der EMA auf verschiedenste Arten in das gesamte Regulierungsverfahren einbezogen werden.
Appendix 2 – Überprüfung der Praxiscodes
Eine Anzahl anerkannter Codes könnte eine wichtige Grundlage für dieses Anleitungsdokument sein.
- Das ECAB Protokoll (eine Beschreibung der Arbeitsvorgänge der ECAB) (European Community Advisory Board, wissenschaftlicher Arbeitsausschuss bei EATG, gegründet 1997))
- Auftrag, Ziele und Verfahrensregeln für die European Medicines Agency Human Scientific Committees’ Working Party with Patients' and Consumers' Organisations (PCWP) (30. Mai 2013)
- Protokoll der EMA Human Scientific Committees’ Working Party with Patients' and Consumers' Organisations (PCWP) (31. Januar 2014)
- 10. Dezember 2009 EMA Überlegungen bezüglich weiterer Patienten- und Konsumentenbeteiligung and den Aktivitäten der Agentur [Reflection Paper on the Further Involvement of Patients and Consumers in the Agency’s Activities]
- EMA Broschüre über die Arbeit mit Patienten und Konsumenten (aktualisiert am 22. April 2015)
- EMA Rahmen der Wechselbeziehungen (überarbeitet am 16. Oktober 2014)
- Empfehlungen vom ECAB Treffen in Bergen, Norwegen 1997
-
EATG ECAB „Der ungeduldige Patient - Vom Zorn zum Aktivismus”Eine systematische Übersicht über die Geschichte, Arbeitsmodelle, Relevanz und Perspektiven des European Community Advisory Board - FDA Patientenvertreterprogramm
- Auf den Patienten konzentrierte Arzneimittelentwicklung bei der FDA; Die Stimme des Patienten: Eine Berichtsserie der auf Patienten konzentrierten Arzneimittelentwicklungs-Initiative bei der FDA
- Auf den Patienten konzentrierte Arzneimittelentwicklung bei der FDA Verbesserung der Nutzen-Risikobewertung bei der regulatorischen Entscheidungsfindung
- WMA Deklaration von Helsinki - Ethische Prinzipien für Medizinische Forschung am Menschen Retrieved 13 July, 2021, from https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/
Quellenangaben
- Aus dem EMA Rahmen übernommen. Europäische Arzneimittel-Agentur [European Medicines Agency] (2014) EMA/637573/2014. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2009/12/WC500018013.pdf. Zuletzt besucht am 21.11.2016.
- Regulation (EC) Nr. 726/2004 http://ec.europa.eu/health/files/eudralex/vol-1/reg_2004_726/reg_2004_726_en.pdf. Zuletzt besucht am 21.11.2016.
*Die Rolle von Konsumenten als Interessengruppe wird im Dialog über das Gesundheitswesen anerkannt. EUPATI konzentriert sich auf Patienten und nicht auf Konsumenten; siehe Trainingsmaterialen und Anleitungsdokumente.