Last update: 3 августа 2015


Введение
В среднем на все исследования и разработки, необходимые для того, чтобы новый лекарственный препарат был доступен для пациентов, уходит более 12 лет и более 1 миллиарда евро.
Разработка лекарственных препаратов — это рисковый бизнес. Большинство разрабатываемых соединений (около 98 %) так и не выходят на рынок. Так происходит потому, что при оценке преимуществ и рисков (негативных побочных эффектов), обнаруживаемых в ходе разработки, сложно сравнивать их с уже имеющимися на рынке препаратами.
Процесс разработки нового лекарственного препарата можно представить в 10 шагах. Следующая статья описывает 7-й шаг. Проверка правильности концепции — клинические исследования 2 фазы
-
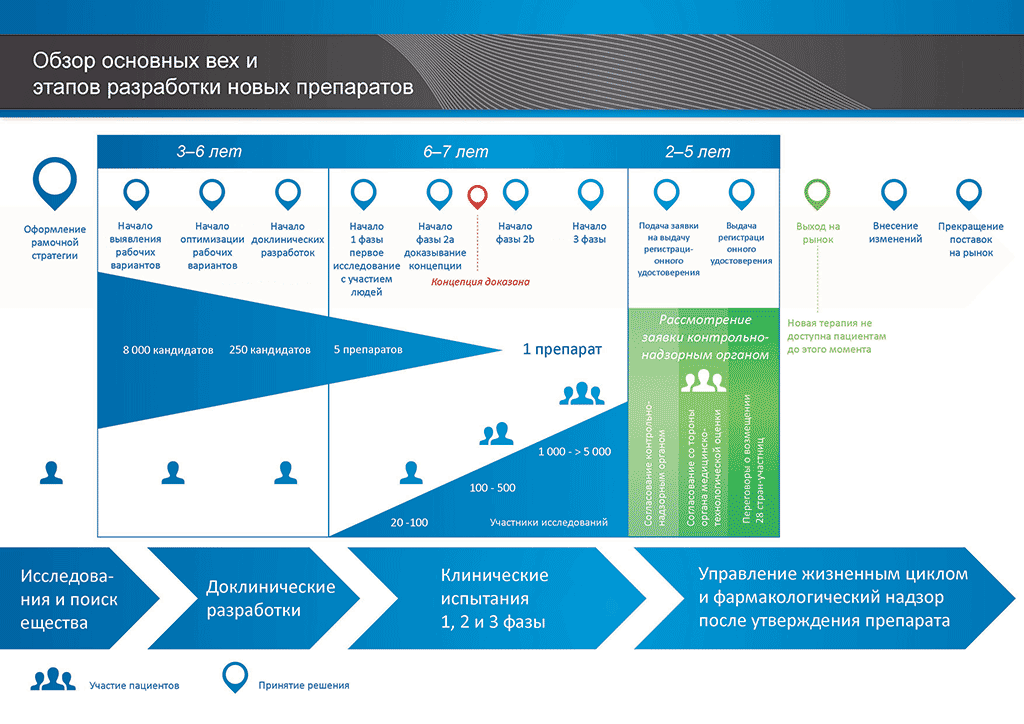
- С момента создания молекулы до момента начала продажи медицинского препарата проходит больше 10 лет, необходимых для тщательнейшего планирования и исследований.
-
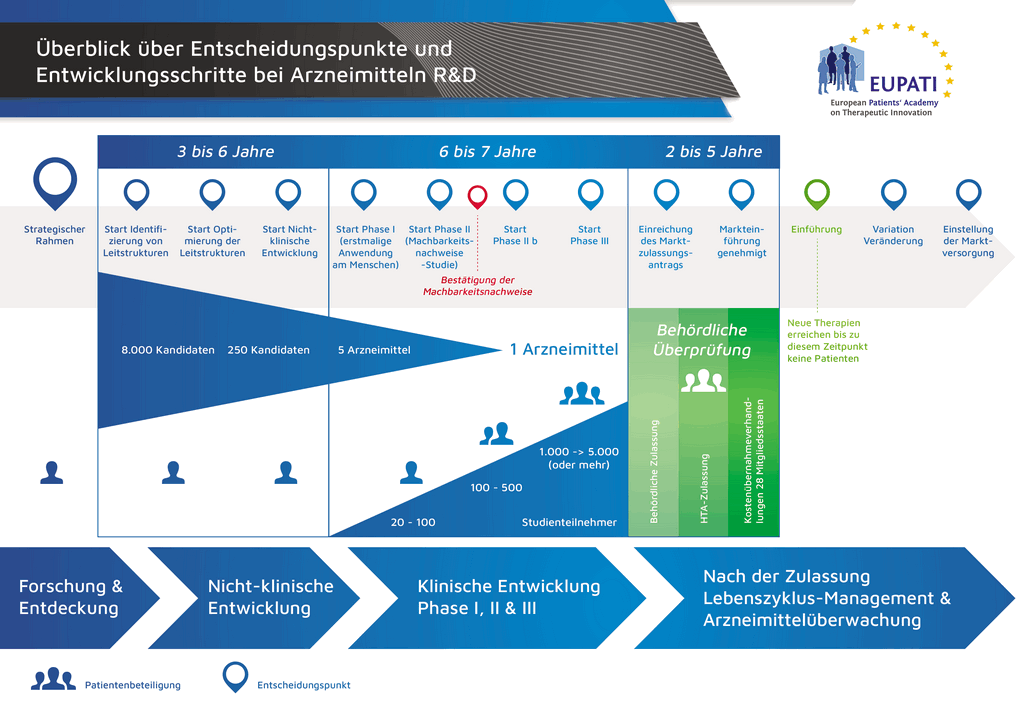
- Es benötigt mehr als 10 Jahre sorgfältiger Planung und Forschung, bis ein Arzneimittel sich vom Molekül zur marktfähigen Behandlung entwickelt hat.
Шаг 7: Проверка правильности концепции — клинические исследования 2 фазы
Исследования на пациентах. После того, как результаты исследований с участием волонтеров показали, что испытания можно продолжать далее, начинаются клинические исследования с участием пациентов с заболеванием, для которого предназначен препарат. При этом применяются те же правила и нормы, что при исследованиях 1 фазы.
В исследованиях 2 и 3 фазы обычно задействованы две группы участников. В одной группе участники получают активное вещество, а в другой — оптимальное известное на текущий момент лечение или неактивное вещество, которое не оказывает никакого влияния на организм («плацебо»). Обычно эти исследования проводятся как «двойные слепые», «рандомизированные» исследования.
- «Двойное слепое» исследование означает, что ни врач, ни участник исследования не знают, кто получает активное вещество, а кто — оптимальное на текущий момент лечение или плацебо.
- «Рандомизированное» исследование означает, что распределение участников между группами лечения производится случайным образом. Обычно это делается с помощью компьютера, генерирующего случайные коды. Ни врач, ни кто-либо еще не могут повлиять на этот процесс.
- «Плацебо-контролируемое» исследование означает, что некоторые участники будут получать плацебо в таких же условиях, что и участники, получающие активное вещество. Это позволяет распознать эффекты препарата. Например, если участник исследования жалуется на головную боль, важно знать, имеет ли это связь с активным веществом. Если в группе плацебо такое же количество участников жалуются на головную боль, то она не может быть вызвана лишь активным веществом.
Все подробности исследования закрепляются в протоколе исследования, а данные фиксируются в индивидуальной регистрационной карте (CRF). Затем результаты оцениваются с использованием статистического анализа.
Эти исследования обычно проводятся с участием 100–500 пациентов. Цель — получить информацию об эффекте препарата непосредственно на заболевание («проверка правильности концепции»). Также именно на этом этапе используются различные дозы препарата с тем, чтобы понять, какая из доз подходит больше всего. Самая удачная доза затем используется на следующем этапе в рамках более масштабных клинических исследований.
Чем больше на этом этапе удается узнать о воздействии препарата на пациентов, тем проще принять решение, стоит ли продолжать исследования. Однако, масштаб исследований 2 фазы слишком мал, чтобы дать достаточный объем сведений об эффективности и безопасности. В этой связи очень важно обнаружить как можно больше данных о действии лекарственного препарата на организм пациентов во избежание рисков неудачи на следующем этапе (в 3 фазе, когда проводится разработка с целью выхода на рынок), который является самым сложным и дорогостоящим в процессе разработки препарата.
Так как исследования 2 фазы проводятся с участием пациентов, эти исследования обычно выполняются в нескольких больничных учреждениях врачами (исследователями), в то время как исследования 1 фазы выполняются в специальных отделениях.
Проводить исследование одновременно в нескольких центрах сложнее, чем в одном центре:
- Все исследователи и медицинский персонал должны быть обучены согласно утвержденному протоколу, благодаря чему исследование проводится аналогичным образом во всех центрах.
- Препарат необходимо экспортировать в различные страны и обеспечить его надлежащее хранение в различных аптечных учреждениях.
- Образцы крови, взятые у участников клинического исследования, обычно отправляются в единую центральную лабораторию.
- Необходимо понимать и выполнять все местные нормы и правила.
- В каждой стране обычно необходимо заключение комитета по этике и согласование со стороны национального компетентного органа.
Все эти действия должна координировать международная команда исследователей.
Резюме: Шаги 1-7
По состоянию на конец исследований 2 фазы программа разработки обычно потребует
- в среднем 8,5 лет и
- в среднем 1 миллиард евро.
Из десяти препаратов, испытанных в исследованиях 1 и 2 фазы, только два (в среднем) препарата перейдут на следующий этап.
Справочная литература
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford: Wiley-Blackwell.
Приложения
- Информационный бюллетень Проверка правильности концепции
Size: 98,527 bytes, Format: .docx
В этом информационном бюллетене описываются клинические исследования 2 фазы, или исследования с целью доказательства механизма действия. Такие исследования проводятся на небольшом количестве пациентов с определенным заболеванием для того, чтобы удостовериться, что исследуемое вещество подействует на это заболевание.
A2-1.02.6-v1.1