

Introduction
Accurate and up-to-date information on medicinal products is essential for both patients and healthcare professionals.
When a person is ill, they may go to their doctor and receive a prescription for medication. The patient might have a number of questions on their mind, such as:
- Can I take the medicine with food?
- Can I take the medicine if I’m pregnant?
- Will this medicine interact with other medicines I’m taking?
These questions will be answered by the package leaflet (PL), a folded leaflet that comes inside the package with the medicine. The content of the PL is governed by European Union legislation, which sets out detailed requirements on medicines information. Directive 2001/83/EC1 sets out the requirements for four different types of information to be provided:
- Labelling on package (immediate and carton)
- Summary of Product Characteristics (SmPC) directed towards healthcare professionals
- Package Leaflet (PL) directed towards the patient
- European Public Assessment Report (EPAR)
There are key requirements in the Directive on how to prepare this information.
Package labelling
The text that appears on the label (inner (immediate) and outer packaging) of medicines is regulated similarly to the package leaflet. The labelling gives information relevant for the patient as well as others in the distribution chain – for instance, the doctor or pharmacist. For the patient, there may be information on special storage conditions – for instance that it should be stored in a refrigerator or protected from light. For the pharmacy, there is information to identify the product in terms of name and active substance, route of administration, etc. In addition, batch numbers are important in case a batch has to be recalled by the manufacturer.
It is important that labelling can be read by all patients. The outer carton of a medicine must include the required information. Labelling on packages must also be both in print and Braille text for those who are visually impaired.
Labelling requirements are harmonised within the EU. However, an individual Member State may decide to additionally include in the label one or more of the following:
- The price of the medicinal product
- The reimbursement conditions
- The legal status – ‘medicinal product subject to medical prescription’ or ‘medicinal product not subject to medical prescription’ (Over the Counter (OTC))
- Authenticity and identification – for instance, bar code.
This additional information is included inside a single boxed area (called the blue Box) located on the outer carton in order to be clearly distinguishable.
Package leaflet (PL)
The pharmaceutical company is required to provide a package leaflet containing all information from the SmPC that is necessary and useful for the patient. The order and content of the PL is strictly regulated. The basic guiding philosophy for the production of a PL is that, after reading it, the patient should fully understand what the medicine is, what it is used for, and how to use it.
The sections of a package leaflet for product X are as follows:
- What X is and what it is used for
- What you need to know before you take (or use) X – for instance, contra-indications, interactions with food or other medicines, precautions
- How to take/use X
- Possible side effects
- How to store X
- Contents of the pack and other information (such as manufacturer and marketing authorisation holder)
The information on potential side effects, or adverse drug reactions (ADRs) is an important element of the PL. At the time of approval of a new medicine, not all ADRs may be known. Rarer ADRs may first occur when larger patient populations are taking the medicine. It is important to involve the patients in the surveillance of new ADRs. Patients have the option of reporting adverse effects to the doctor or to the pharmacy. In many EU member states, patients may even report directly to the regulatory authorities.
The pharmacovigilance legislation requires that patients be informed on the importance of reporting new ADRs. A standard text must appear in all PLs, as shown below:
“If you get any side effects, talk to your doctor, pharmacist, or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly via the national reporting system. By reporting side effects, you can help provide more information on the safety of this medicine.”
Involving patients in the review of the package leaflet
The company should consult with appropriate patient groups to ensure the product information (both content and layout) is legible, clear, and easy to use so that patients can locate important information within the package leaflet, understand it, and act appropriately. One way to do this is by performing a ‘user testing’ of the package leaflet. For this purpose, the European Commission has issued a Guideline on the readability of the labelling and package leaflet of medicinal products for human use.2
Summary of Product Characteristics
In addition to the labelling and PL, the company must provide a Summary of Product Characteristics (SmPC), which is an essential document in the approval process for a new medicine. This document should include all necessary information in a condensed form for healthcare professionals.
The SmPC contains the necessary information needed by healthcare professionals in order to inform the patients on the benefits and the risks of the chosen medicine. Furthermore, detailed guidance on the use is included along with relevant warnings on what to do and what to avoid during the medication period. The SmPC is kept up-to-date with current information on the adverse reactions, safety, and benefit-risk balance of the medication.
-
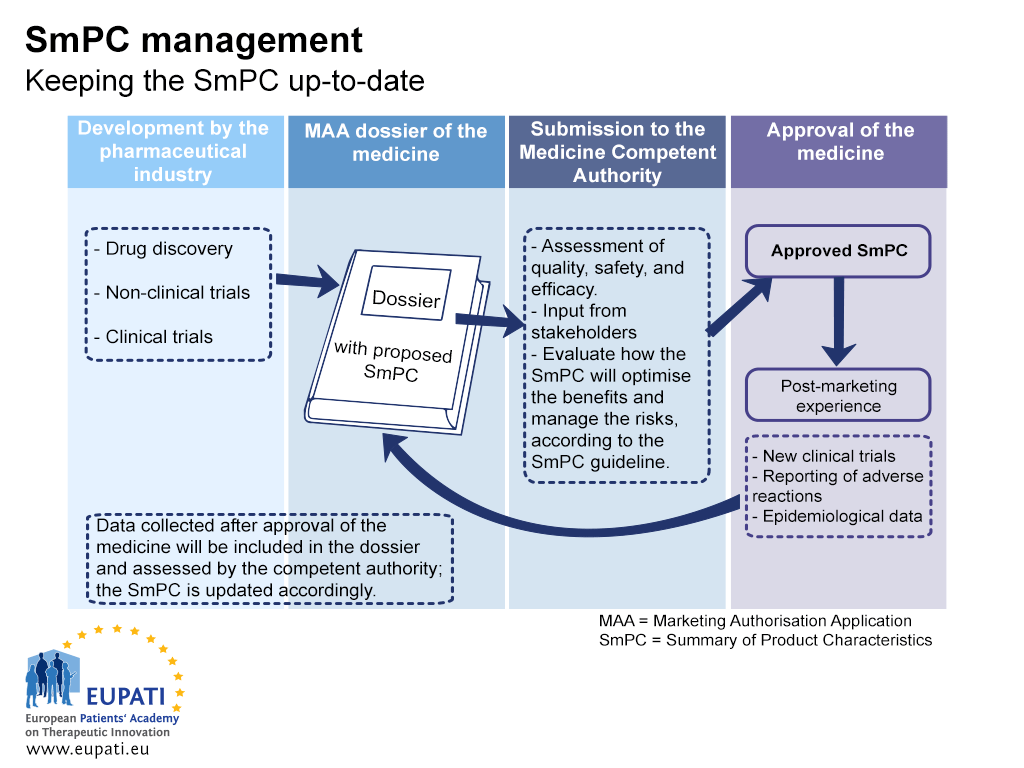
- The Summary of Product Characteristics (SmPC) must be kept up-to-date throughout the lifecycle of a medicine.
European Public Assessment Reports (EPARs)
An EPAR provides public information on a medicine, including how it was assessed together with a ‘public-friendly’ summary. The intention is to provide transparent and useable information.
These public documents contain a summary of the information contained within the dossier and the result of the assessment carried out by the competent authorities. However, some of the information is regarded as confidential and is not included – typically, this includes detailed information on the manufacturing of the products. EPARs are updated periodically to reflect the latest regulatory information on medicines.
Changes in information on medicinal products
Relevant content in the SmPC and the PL cannot be changed without submitting a ‘variation application’. Such changes can be initiated by the company as well as the regulatory authorities – for instance, in case of new knowledge on product safety or efficacy. Regulatory initiatives may arise from what are known as ‘referrals’. Referrals may be initiated by EU Member States or the Commission, for instance, and involve scientific considerations in EMA committees.
A new medicinal product should be closely monitored after it becomes available to patients. As mentioned above, larger patient populations are exposed to the medicine only when it is on the market. Accordingly, rarer ADRs may only be spotted after marketing. Both the regulatory agencies and the pharmaceutical companies collect ADR reports. Based on the post-authorisation information compiled in Periodic Safety Update Reports (PSURs), the benefit-risk balance is regularly re-considered and re-assessed. Any new safety information resulting from these re-assessments can lead to updates of the SmPC and the PL.
After ADR reporting, the regulators may want to initiate a safety referral to re-assess the benefit-risk balance of the medicinal product and to update the product information when needed. This could be for new or older products. In such cases, a harmonised EU action is important. All patients and doctors should have access to any new information on benefits or risks.
In the case of new and serious ADRs, it may be that urgent action is required. One possibility in such cases is to start the ‘Urgent Union Procedure’. This procedure may be initiated by an EU Member State or by the Commission. The result can be major changes in the conditions for marketing authorisation, the suspension or even the revocation of the marketing authorisation. Most often, this kind of urgent union safety procedure will generate a safety communication published by the EMA, and the update of the product information of the medicinal product(s) affected.
Patient involvement in information on medicinal products
As described above, a lot of information on medicines is directed specifically towards patients.
The EMA invites patients and consumer representatives to review information on medicines published by them, such as public-friendly summaries of EPARs and PLs. The purpose is to have feedback on whether the information is readable and understandable for the target population.
Further Resources
- European Medicines Agency (2014). EMA/403106/2014 Mandate and objectives for the EMA working party on Quality Review of Documents (QRD). Retrieved 6 July, 2021, from https://www.ema.europa.eu/en/documents/presentation/presentation-summary-product-characteristics_en.pdf
- European Medicines Agency (2012). Draft presentation: Summary of product characteristics. Retrieved 6 July, 2021, from https://www.ema.europa.eu/en/documents/presentation/presentation-summary-product-characteristics_en.pdf
- European Medicines Agency (2022). EMA/649909/2021 Adopted Engagement framework: European Medicines Agency and patients, consumers, and their organisations. Retrieved 12 February, 2024, from https://www.ema.europa.eu/en/documents/other/engagement-framework-european-medicines-agency-and-patients-consumers-and-their-organisations_en.pdf
- European Medicines Agency (2014). EMA/652164/2014 Annex II: EMA activities where patients and consumers are involved. Retrieved 11 September, 2015 from https://www.ema.europa.eu/en/partners-networks/patients-and-consumers/getting-involved
- European Medicines Agency (2013). EMA/413422/2013 – rev 1. Incorporating patients’ views during evaluation of benefit-risk by the EMA Scientific Committees. Retrieved 11 September, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Other/2014/09/WC500173508.pdf
- European Medicines Agency. (n.d.). Patients and Consumers Working Party. Retrieved 12 February, 2024, from https://www.ema.europa.eu/en/committees/working-parties-and-other-groups/comp-working-parties-and-other-groups/patients-and-consumers-working-party
- European Medicines Agency (2006). EMEA/126757/2005 Reflection paper: EPAR summary for the public. Retrieved 11 September, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/10/WC500004173.pdf
Attachments
- Presentation: Information on Medicinal Products
Size: 547,126 bytes, Format: .pptx
A presentation describing the information in medicinal products, which can be adapted for own use.
A2-5.12-v1.1