Wnioski o wydanie pozwolenia na dopuszczenie do obrotu
Wprowadzenie
Proces rozwoju leku to długa podróż. Ostatecznym celem każdego procesu rozwoju jest uzyskanie zgody na wprowadzenie na rynek nowego produktu leczniczego — pozwolenia na dopuszczenie do obrotu (MA). W firmach farmaceutycznych działy rejestracji są (lub powinny być) integralną częścią wszystkich procedur w całym cyklu życia produktu leczniczego. Dział rejestracji jest odpowiedzialny w szczególności za wnioski, które muszą być złożone przed każdym badaniem klinicznym, za opracowanie i złożenie dossier do wniosku o wydanie pozwolenia na dopuszczenie do obrotu (MAA) oraz za inne działania po przyznaniu MA, na przykład za złożenie wniosku o wprowadzenie zmiany do pozwolenia na dopuszczenie do obrotu. Specjaliści ds. rejestracji muszą posiadać gruntowną wiedzę na temat wszystkich obowiązujących regulacji dotyczących leków oraz całego procesu rozwoju.
-
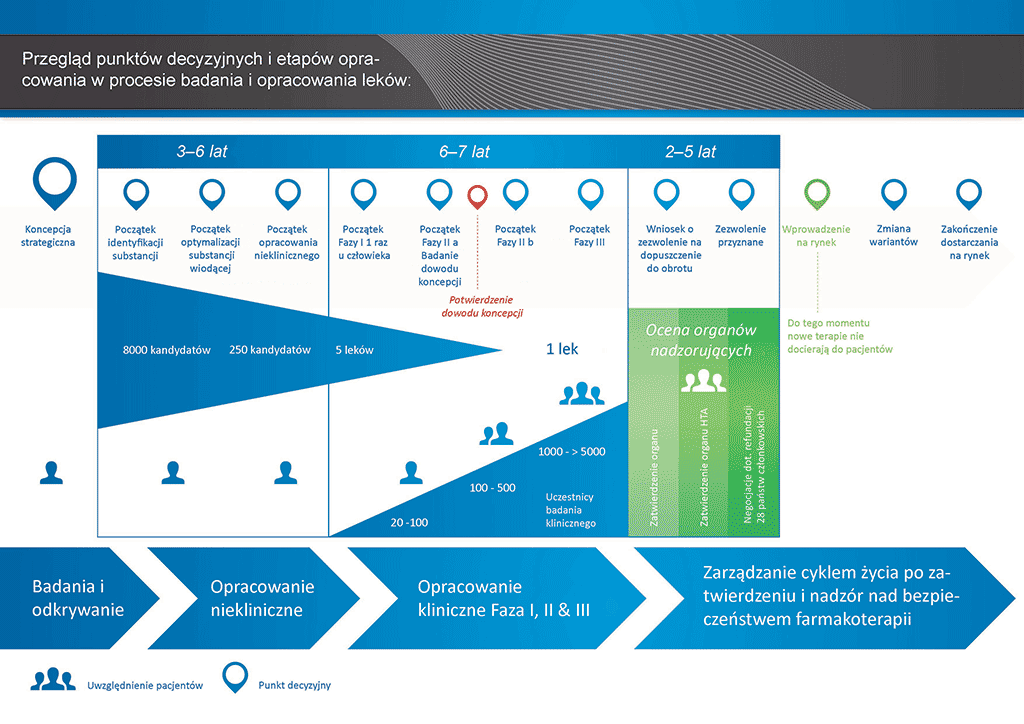
- Proces opracowania leku zajmuje ponad dziesięć lat planowania i badań pozwalających przejść od cząsteczki do dostępnego na rynku leczenia.
Rycina 1 — Ogólny zarys procesu rozwoju leków
Składanie wniosków o wydanie pozwolenia na dopuszczenie do obrotu
Firma farmaceutyczna musi na wczesnym etapie prac rozwojowych zdecydować, jaki typ wniosku o wydanie pozwolenia na dopuszczenie do obrotu będzie składać, np.:
- Wniosek pełny — zob. trójkąt wspólnego dokumentu technicznego (CTD) poniżej.
- Wniosek uproszczony (wniosek ograniczony).
- Wniosek bibliograficzny — oparty na istniejącej literaturze naukowej.
Wnioski wymagają złożenia dossier do odpowiednich władz. Rycina 1 przedstawia procesu rozwoju nowego innowacyjnego leku. Ten rodzaj leku wymaga złożenia pełnego dossier, które musi zawierać wszystkie elementy dokumentacji leku.
Jakie elementy zawiera dossier?
Na rycinie 2 pokazano elementy składające się na wspólny dokument techniczny (CTD) — dossier składane do organów regulacyjnych jako wniosek o wydanie pozwolenia na dopuszczenie do obrotu (MAA) w Kanadzie, Europie, Japonii, Szwajcarii, Stanach Zjednoczonych i innych krajach. Format CTD został opracowany przez Międzynarodową Konferencję ds. Harmonizacji Wymagań Technicznych dla Rejestracji Produktów Leczniczych Stosowanych u Ludzi (ICH). Dokument CTD jest stosowany przy wszystkich typach MAA w UE niezależnie od rodzaju procedury (procedura zcentralizowana (ang. centralised procedure, CP), wzajemnego uznania (ang. mutual recognition procedure, MRP), zdecentralizowana (ang. decentralised procedure, DCP) lub narodowa (ang. national procedure, NP)) i typu wniosku (autonomiczny, dla leku generycznego itd.). Format CTD ma zastosowanie do wszystkich typów produktów (nowe substancje chemiczne, radiofarmaceutyki, szczepionki, preparaty ziołowe itp.).
CTD składa się z pięciu odrębnych modułów.
- Moduł 1: „Regionalne informacje administracyjne”.
- Moduł 2: „Podsumowania i przeglądy”.
- Moduł 3: „Jakość”.
- Moduł 4: „Sprawozdania z badań nieklinicznych”.
- Moduł 5: „Sprawozdania z badań klinicznych”.
Moduły CTD od 2 do 5 są wspólne dla wszystkich regionów, natomiast moduł 1 jest inny w każdym regionie i nie jest uznawany za część CTD. Większość dokumentacji dotyczącej jakości, bezpieczeństwa i skuteczności leku jest zawarta w modułach od 3 do 5. Specjaliści ds. rejestracji zapewniają zgodność składanej dokumentacji z odpowiednimi rozporządzeniami, dyrektywami i wytycznymi. Tworzą oni również podsumowania, które są zamieszczane w module 2.
Rycina 2: Trójkąt wspólnego dokumentu technicznego.
Moduł 1: Regionalne informacje administracyjne
Moduł 1 CTD zawiera wszystkie informacje administracyjne niezbędne na poziomie regionalnym. UE ma własną wersję modułu 1. Składa się on z następujących 10 elementów:
1.0 Pismo przewodnie
1.1 Szczegółowy spis treści
1.2 Formularz wniosku
1.3 Druki informacyjne
Są to informacje, z których będą korzystać zarówno pracownicy ochrony zdrowia jak i pacjenci. Obejmują one charakterystykę produktu leczniczego (ChPL) będącą szczegółowym dokumentem przeznaczonym dla pracowników ochrony zdrowia, oznakowanie oraz ulotkę dla pacjenta. Konieczne jest przeprowadzenie badania czytelności ulotki w celu wykazania, że jest ona zrozumiała dla laików. Odnośne władze krajowe oraz EMA opublikowały szablony we wszystkich językach UE, aby szczegółowo przestawić format i treść druków informacyjnych.
1.4 Informacje dotyczące ekspertów
Moduł 2 CTD zawiera podsumowania i przeglądy sporządzone przez ekspertów. Każdy z tych ekspertów powinien dostarczyć curriculum vitae (CV) i podpisać oświadczenie potwierdzające, że podczas tworzenia podsumowań przestrzega zasad określonych w odpowiednich rozporządzeniach lub dyrektywach.
1.5 Szczegółowe wymagania dla różnych typów wniosków
Dodatkowe informacje wymagane w przypadku szczególnych rodzajów wniosków takich jak wnioski bibliograficzne, wnioski dla leków generycznych, wnioski hybrydowe i wnioski dla leków biopodobnych, wnioski o (rozszerzony) okres wyłącznego prawa do danych / wyłączności rynkowej, wnioski w wyjątkowych okolicznościach lub wnioski o warunkowe pozwolenie na dopuszczenie do obrotu.
1.6 Ocena ryzyka dla środowiska naturalnego
Wszystkie substancje czynne w lekach mogą stwarzać potencjalne ryzyko dla środowiska naturalnego, a wszystkie substancje lub ich metabolity ostatecznie trafiają do środowiska naturalnego. Firma musi odnieść się do możliwego wpływu użytkowania, przechowywania i usuwania leku na środowisko naturalne.
1.7 Informacja dotycząca wyłączności rynkowej dla sierocego produktu leczniczego
Jeśli lek jest określony jako sierocy produkt leczniczy przeznaczony do leczenia choroby rzadkiej, wymagane są specjalne informacje. Jeśli inny lek będący w obrocie ma już status wyłączności rynkowej dla takiego samego wskazania, nowy lek może zostać dopuszczony wyłącznie na warunkach specjalnych.
1.8 Informacje dotyczące systemu nadzoru nad bezpieczeństwem farmakoterapii
Należy zamieścić opis systemu nadzoru nad bezpieczeństwem farmakoterapii i systemu zarządzania ryzykiem. Wnioskodawca musi wykazać, że posiada odpowiedni system nadzoru nad działaniami niepożądanymi i potencjalnym ryzykiem. Obejmuje to przedstawienie dowodu, że wnioskodawca dysponuje usługami osoby wykwalifikowanej odpowiedzialnej za nadzór nad bezpieczeństwem farmakoterapii oraz że posiada niezbędne środki dla powiadamiania o wszelkich działaniach niepożądanych, które wystąpiły w UE albo w państwie trzecim (art. 8(n) dyrektywy 2001/83/WE).
1.9 Informacje dotyczące badań klinicznych
Wnioskodawca musi przedstawić oświadczenie stwierdzające, że wszystkie badania kliniczne nad lekiem przeprowadzone poza UE spełniają wymagania obowiązujące w UE.
1.10 Informacje dotyczące produktów leczniczych stosowanych w pediatrii
W UE wszystkie nowe leki muszą być brane pod uwagę jako leki do stosowania u dzieci i młodzieży. Ogólnie rzecz biorąc, powinny być przebadane u dzieci. Jednak można uzyskać zwolnienie z tego wymogu, jeśli choroba występuje tylko u osób starszych lub dorosłych lub jeśli lek prawdopodobnie będzie nieskuteczny lub niebezpieczny dla niektórych lub wszystkich dzieci i młodzieży. Jeśli zwolnienie nie zostanie przyznane, firma musi opracować plan badań pediatrycznych (PIP), chyba że zostanie przyznane odroczenie, a w takim przypadku plan PIP można opracować w późniejszym terminie. W tej części powinna znaleźć się kopia zwolnienia lub decyzji dotyczącej PIP (w tym odroczenie, jeśli ma zastosowanie).
Jaką postać ma dossier?
W większości przypadków składanie dossier w postaci papierowej nie jest już możliwe, co oznacza, że cała dokumentacja w pięciu modułach CTD powinna mieć ustandaryzowany format elektroniczny: eCTD. eCTD nie jest zwykłym zbiorem dokumentów PDF czy też jednym dużym plikiem PDF. Jest to raczej standard szczegółowo opisujący strukturę folderów i plików, którą należy zachować, aby firma oraz organy regulacyjne mogły łatwo poruszać się po dossier, tak jak po normalnym katalogu komputerowym.
Jak wygląda proces składania wniosku?
Przed złożeniem wniosku wnioskodawca musi dokładnie rozważyć wszystkie kwestie logistyczne i regulacyjne. Obejmuje to wybór procedury uzyskiwania pozwolenia na dopuszczenie do obrotu, zgodnie z którą składany będzie wniosek: procedura zcentralizowana (CP), procedura wzajemnego uznania (MRP), procedura zdecentralizowana (DCP) lub procedura narodowa (NP).
-
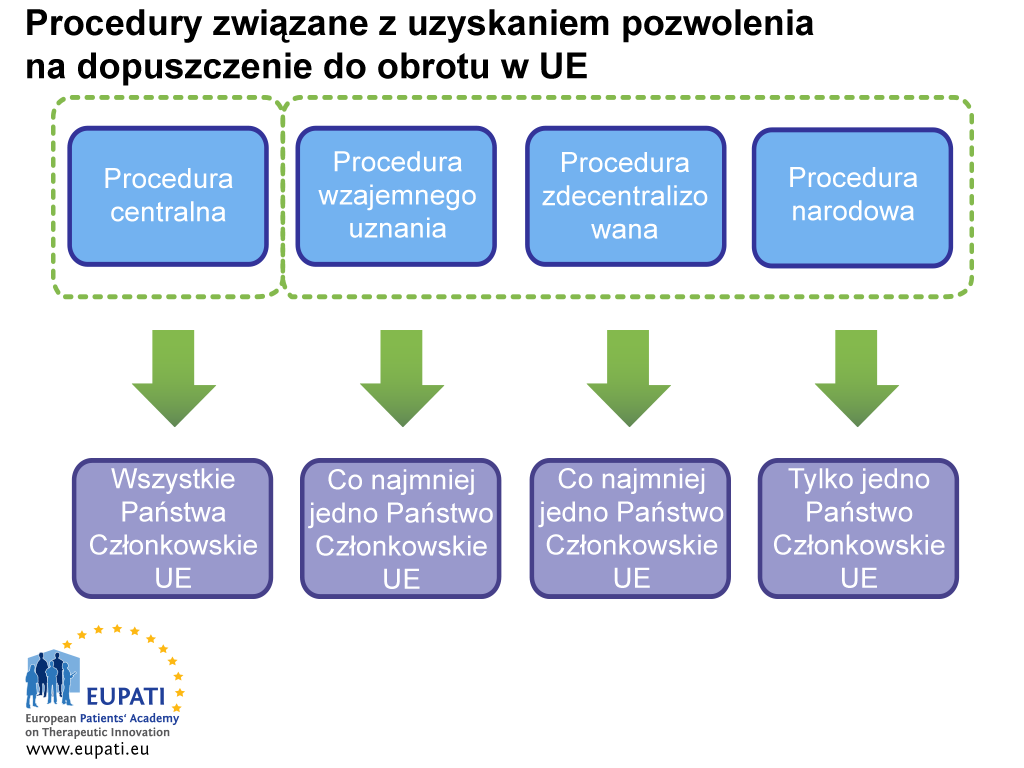
- W uzyskanie pozwolenia na dopuszczenie leku do obrotu zaangażowane są różne strony zależnie od procedury wybranej (lub obowiązującej) sponsora.
Odpowiedzi na wiele pytań dotyczących wniosków składanych na drodze procedury CP można znaleźć na witrynie internetowej EMA.
Spotkania poprzedzające złożenie wniosku
Spotkania firmy z przedstawicielami organów regulacyjnych poprzedzające złożenie wniosku odbywają się sześć do siedmiu miesięcy przed datą złożenia wniosku. Spotkania te są organizowane po to, aby firma mogła zwrócić się o dalsze informacje i wytyczne przed zakończeniem opracowywania dossier.
W przypadku wniosku składanego na drodze procedury centralnej zespół odpowiedzialny za projekt z ramienia firmy spotyka się z zespołem EMA, który będzie zaangażowany w ocenę wniosku. W przypadku procedur MRP, DCP lub NP spotkania poprzedzające złożenie wniosku z odpowiednimi odnośnymi władzami krajowymi są możliwe i w równym stopniu przydatne.
Składanie wniosku o wydanie pozwolenia na dopuszczenie do obrotu (MAA)
W przypadku procedury CP wniosek można składać wyłącznie do EMA w formacie eCTD, chyba że wyrażono zgodę na wyjątek. Dokument eCTD jest składany przez portal internetowy.
W przypadku wniosków składanych na drodze procedur MRP, DCP lub NP sytuacja jest bardziej skomplikowana. Te wnioski mogą być składane do 31 różnych agencji. Sieć HMA (sieć szefów agencji leków oparta na współpracy między państwami członkowskimi) umożliwia obecnie korzystanie z rozwiązania podobnego do oferowanego przez EMA i określanego jako wspólna europejska platforma składania wniosków (ang. Common European Submission Platform, CESP). Dzięki CESP firma wysyła dossier do systemu tylko raz; wszystkie zaangażowane państwa członkowskie mogą następnie pobrać złożony wniosek z repozytorium CESP. Platforma ta umożliwia tez komunikację między agencjami a wnioskodawcą.
Etap weryfikacji
Kiedy EMA lub odnośne władze krajowe otrzymają MAA, dossier jest najpierw weryfikowane w celu sprawdzenia, czy załączono wszystkie niezbędne dokumenty. W razie pytań wnioskodawca ma możliwość dostarczenia koniecznych odpowiedzi i dokumentacji na ich potwierdzenie. Po zweryfikowaniu MAA rozpoczyna się ocena.
A2-5.11-V1.1