Vergunning voor het in de handel brengen
Inleiding
Alle geneesmiddelen moeten een vergunning voor het in de handel brengen hebben om legaal op de markt te kunnen worden gebracht in de Europese Economische Ruimte (EER).1 Het uiteindelijke doel van vergunningverlening is ervoor te zorgen dat veilige, effectieve en hoogwaardige geneesmiddelen snel beschikbaar kunnen komen voor burgers van de EER. Hierbij dient te worden aangemerkt dat voor medische hulpmiddelen andere procedures gelden dan voor overige geneeskundige producten voordat ze in de handel verkrijgbaar kunnen worden gemaakt.
Nationale bevoegde autoriteiten (kortweg ‘toezichthoudende instanties’ zijn nationale overheidsinstanties die verantwoordelijk zijn voor de beoordeling van vergunningaanvragen en het verlenen van handelsvergunningen voor geneeskundige producten die op hun markt worden geplaatst via nationale, gedecentraliseerde, of wederzijdse erkenningsprocedures. Het Europees Geneesmiddelenbureau (EMA) in Londen is verantwoordelijk voor de coördinatie van de wetenschappelijke beoordeling van de aanvragen voor Europese handelsvergunningen voor geneeskundige producten onder de gecentraliseerde procedure (centralised procedure, CP). De beoordeling wordt uitgevoerd door deskundigen van bevoegde nationale autoriteiten. Na positieve beoordeling verleent de Europese Commissie de handelsvergunning.
Procedures bij vergunningverlening
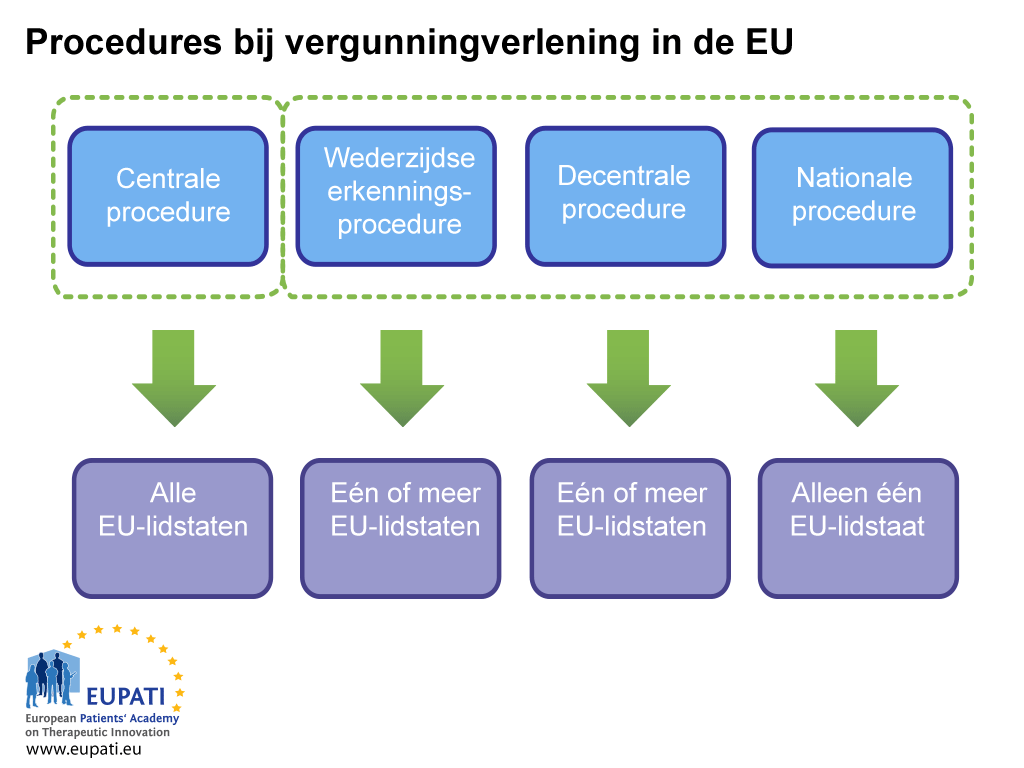
Er zijn twee manieren om een handelsvergunning te verkrijgen: via de gecentraliseerde procedure of via een niet-gecentraliseerde procedure. Deze laatste behelst de gedecentraliseerde procedure (decentralised procedure, DCP), de procedure van wederzijdse erkenning (mutual recognition procedure, MRP) en de national procedure. Elk systeem heeft eigen juridische bepalingen en verantwoordelijkheden voor de bevoegde autoriteit (EMA of nationale instanties) en vergunninghouders.
De gecentraliseerde procedure (CP)
Voor de meeste geneesmiddelen is deze route van toepassing. Industrie dient een enkele aanvraag in bij het Europees Geneesmiddelenbureau (EMA). Het Comité voor geneesmiddelen voor menselijk gebruik (CHMP) van het EMA – dat bestaat uit één door elk van de EU-lidstaten en EER-landen aangewezen lid en vijf deskundige wetenschappers – adviseert de Europese Commissie of een geneesmiddel moet worden goedgekeurd. Als een geneesmiddel op deze manier wordt goedgekeurd, wordt een vergunning voor het in de handel brengen verleend voor alle EU- en EER-landen. Deze wetenschappelijke beoordelingsprocedure duurt maximaal 210 werkdagen, maar deze telling kan worden stopgezet (een ‘clock stop’).
Gedecentraliseerde procedure, de procedure van wederzijdse erkenning
Bij deze procedures worden handelsvergunningen aangevraagd voor meerdere lidstaten tegelijkertijd, waarbij één land de leiding neemt als rapporterende lidstaat (‘reference member state’, RMS). Als de indiening succesvol is, wordt het geneesmiddel goedgekeurd voor het in de handel brengen in de rapporterende lidstaat en de andere betrokken lidstaten (‘concerned member state’, CMS). Deze procedures zijn gebaseerd op het principe van erkenning van de beoordeling van de rapporterende lidstaat door de betrokken lidstaten. De meeste generieke geneesmiddelen worden goedgekeurd via de gedecentraliseerde procedure.
Nationale procedure
Onafhankelijke nationale procedures zijn strikt beperkt tot geneesmiddelen die slechts in één lidstaat moeten worden goedgekeurd en in de handel gebracht. Deze procedure wordt tegenwoordig nauwelijks gevolgd voor nieuwe producten.
Een procedure voor vergunningverlening selecteren
Een farmaceutisch bedrijf is normaal gesproken vrij om zelf te beslissen welke procedure het gaat gebruiken. Bij dit zakelijke besluit kunnen verschillende redenen een rol spelen om voor een bepaalde procedure te kiezen. De CP is verplicht voor:
- producten afkomstig van biotechnologische processen,
- geneesmiddelen voor geavanceerde therapie (zoals gentherapie, somatische celtherapie of geneesmiddelen gemaakt met behulp van weefseltechnologie)
- weesgeneesmiddelen,
- geneesmiddelen bedoeld voor de behandeling van hiv of aids, kanker, neurodegeneratieve stoornissen, auto-immuun- en andere immuundisfuncties, virusziekten of diabetes.
De gecentraliseerde procedure kan ook worden gebruikt voor geneesmiddelen die een belangrijke therapeutische, wetenschappelijke of technische innovatie zijn of die in het belang van de volksgezondheid zijn, zoals geneesmiddelen voor gewichtsverlies.
Als een bedrijf een bestaand geneesmiddel wil ontwikkelen met een handelsvergunning voor een andere indicatie, dan moet meestal een geheel nieuwe handelsvergunning worden aangevraagd.
-
- Er zijn verschillende actoren betrokken bij het verlenen van een vergunning voor het in de handel brengen van een geneesmiddel, afhankelijk van de procedure die de sponsor wil (of moet) volgen.
Wetenschappelijke beoordeling van geneesmiddelen
Elk geneesmiddel moet een standaard ‘dossier’ hebben voor beoordeling van de aanvraag van een vergunning voor het in de handel brengen, ongeacht de procedure. Het gemeenschappelijke technische document (‘common technical document’, CTD) is een internationaal erkend geharmoniseerd schema dat moet worden gevolgd voor aanvragen die zijn bedoeld voor indiening bij toezichthoudende autoriteiten. Het CTD bestaat uit een aantal documenten waarin de aanvrager aantoont dat het geneeskundige product de vereiste kwaliteit heeft en dat het veilig en werkzaam is.
Afwijzing van de aanvraag van de handelsvergunning
Een handelsvergunning wordt niet verleend wanneer het volgende van toepassing is op het geneeskundige product:
- Baten-risicobalans wordt niet beschouwd als gunstig, of
- Therapeutische werkzaamheid is onvoldoende onderbouwd door de aanvrager, of
- Kwalitatieve en kwantitatieve samenstelling is niet zoals opgegeven.
Validiteit handelsvergunning en opvolging
De handelsvergunning wordt na vijf jaar vernieuwd op grond van een herbeoordeling van de baten-risicobalans door de bevoegde autoriteit van de lidstaat die de vergunning verleent. Eenmaal vernieuwd is de handelsvergunning voor onbeperkte tijd geldig, tenzij de bevoegde autoriteit besluit (gerechtvaardigd op grond van geneesmiddelenbewaking) om een extra verlenging van vijf jaar in te stellen.
Alle houders van handelsvergunningen zijn verplicht om gegevens op het gebied van veiligheidsopvolging te verstrekken en zich te houden aan andere vereisten na vergunningverlening. Eenmaal in de handel gebracht moeten geneesmiddelen in feite continu worden bewaakt (geneesmiddelenbewaking of farmacovigilantie). Dit is om ervoor te zorgen dat eventuele aspecten die van invloed zouden kunnen zijn op het veiligheidsprofiel van een geneesmiddel worden ontdekt en beoordeeld en dat de nodige maatregelen worden getroffen.
Als het geneesmiddel in de handel is, kunnen er nieuwe gegevens of kan er nieuwe informatie over de werkzaamheid, veiligheid of productie beschikbaar komen. In dat geval is het de taak van de vergunninghouder om een aanvraag in te dienen voor een ‘wijziging’ van een geldige handelsvergunning bij de bevoegde autoriteit(en) in alle lidstaten die het geneeskundige product eerder hebben goedgekeurd.
Samenwerking met patiënten bij vergunningverlening
Samenwerking met patiënten bij vergunningverlening is van oudsher zeer beperkt. Sommige bevoegde autoriteiten denken er echter over na hoe ze patiënten kunnen betrekken bij procedures voor vergunningverlening – bijvoorbeeld via patiëntenraadpleging en/of -vertegenwoordiging in de Commission on Human Medicines binnen het Medicines and Healthcare products Regulatory Agency (MHRA) in Groot-Brittannië. Patiënten zijn al betrokken bij besprekingen van baten en risico’s bij het EMA als leden van het ‘Pharmacovigilance Risk Assessment Committee’ (PRAC), via hun deelname aan procedures inzake wetenschappelijk advies/protocolassistentie, en in bijeenkomsten van de Scientific Advisory Group (SAG). Het CHMP is ook een proefproject gestart waarbij patiënten aanwezig zijn tijdens de mondelinge uiteenzettingen voorafgaand aan de besluitvorming.
Behartigers van patiëntenbelangen kunnen ook invloed uitoefenen op nationale/Europese wetgeving rondom het verlenen van handelsvergunningen.
Referenties
- The European Economic Area (EEA) unites the EU Member States and the three EEA EFTA States (Iceland, Liechtenstein, and Norway) into the internal market governed by the same basic rules. These rules aim to enable goods, services, capital, and persons to move freely about the EEA in an open and competitive environment, a concept referred to as the four freedoms.
Bijlagen
- Vergunning voor het in de handel brengen
Size: 480,114 bytes, Format: .pptx
Meer informatie over de procedures voor het verwerven van een handelsvergunning voor een geneeskundig product in de Europese Economische Ruimte (EER).
A2-5.07-v1.1