Types d’études réalisées dans le cadre du développement clinique précoce
Introduction
Les études généralement réalisées dans le cadre du développement clinique précoce (phase I et phase II) ont plusieurs objectifs. Ces essais cliniques précoces visent surtout à établir qu’un médicament expérimental est sûr chez l’homme. Elles cherchent également à prouver l’efficacité de ce médicament contre la maladie ou le problème ciblé(e).
Une réponse doit être apportée aux questions essentielles suivantes durant le développement clinique précoce :
- Phase I
- Le médicament est-il sûr pour les humains ? À quels niveaux ? (Tolérance)
- Quelle est l’action de l’organisme sur le médicament ? (Données pharmacocinétiques (PK))
- Quelle est l’action du médicament sur l’organisme ? (Données pharmacodynamiques (PD))
- Quelles interactions existent à ce niveau ? (Interactions avec d’autres médicaments, interactions alimentaires, etc.)
- Le médicament est-il actif ?
- Phase II
- Le médicament est-il sûr pour les patients ? (Sécurité)
- Quelle est l’action du médicament sur l’organisme ? (Données pharmacodynamiques (PD))
- Le médicament semble-t-il être efficace chez les patients ? À quelle(s) dose(s) ? (Effet)
- Comment les essais confirmatoires doivent-ils être conçus ? (Critères d’évaluation, population cible, autres prises de médicaments (concomitantes), etc.)
Le développement des médicaments est généralement représenté comme une suite chronologique de phases, pourtant les études réalisées lors de chaque phase sont plutôt organisées en fonction de leurs objectifs. Comme l’indique le diagramme ci-dessous, les études qui appartiennent à une phase précoce peuvent être menées plus tard dans le processus de développement d’un médicament, au fur et à mesure de l’émergence de données suggérant la nécessité d’informations supplémentaires.
Études à dose unique croissante/à doses multiples croissantes
Les études à dose unique croissante et les études à doses multiples croissantes sont généralement les premières études à être menées chez l'humain.
Objectifs des études
Les études à dose unique croissante et les études à doses multiples croissantes :
- Analysent la sécurité et la tolérance
- Identifient la dose maximum tolérée (DMT)
- Déterminent les caractéristiques pharmacocinétiques (PK) générales
- Étudient la façon d'atteindre une concentration stable du médicament dans l'organisme sur la durée. Ces conditions portent le nom de paramètres à l'état d'équilibre (dépendance temporelle de l'accumulation).
- Réalisent une exploration préliminaire de l'excrétion par l'organisme (identification de métabolites)
Méthodologie des études
La dose de départ des études à dose unique croissante et des études à doses multiples croissantes est déterminée à partir des résultats des études toxicologiques non cliniques. Cette dose est ensuite augmentée selon les schémas posologiques présentés dans les documents d'application de la réglementation. Les études prennent fin conformément à des règles d'arrêt, qui tiennent compte de la toxicité ou de l'absence de toxicité (exposition maximisée, données pharmacodynamiques maximisées, etc.).
Études de bilan de masse : Absorption, Distribution, Métabolisme, Excrétion (ADME)
Objectifs des études
Les études ADME ont pour objectif de comprendre et de caractériser le profil pharmacocinétique (PK) du médicament expérimental, c'est-à-dire, l'action de l'organisme sur le médicament. Ces études analysent la manière dont le médicament est absorbé, réparti, métabolisé, puis excrété par l'organisme (voir l'image ci-dessous).
#mla_gallery-2 { margin: auto; width: 100%; } #mla_gallery-2 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-2 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-2 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
-
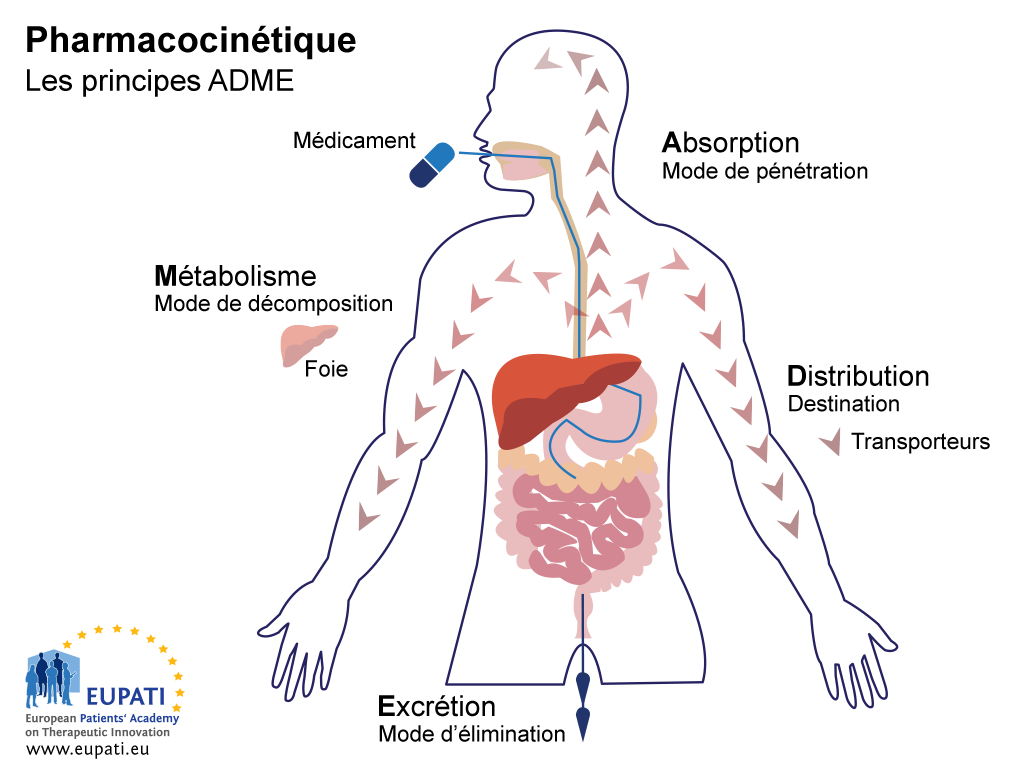
- Les principes clés de la pharmacocinétique (l’étude des effets de l’organisme sur un médicament) sont représentés par l’acronyme ADME.
Méthodologie des études
Les études ADME prévoient généralement l'utilisation d'une seule dose du médicament sur un petit nombre (souvent quatre à six) de volontaires sains de sexe masculin (pour ne faire courir aucun risque aux femmes en âge de procréer), selon la voie d'administration prévue. Ces études sont souvent associées à la phase I du développement, mais elles peuvent être réalisées tout au long du développement d'un produit médicamenteux.
Les études ADME fournissent également des informations sur la biodisponibilité, c'est-à-dire la fraction de la dose administrée du principe actif qui atteint la circulation sanguine.
Études de biodisponibilité et de bioéquivalence
Objectifs des études
Les études de biodisponibilité évaluent le taux et l'étendue d'absorption d'un médicament. Elles analysent la concentration du principe actif dans la circulation sanguine dans le temps.
Les médicaments administrés par des voies différentes ont des profils de biodisponibilité différents. Par exemple, les médicaments administrés directement dans la circulation sanguine par injection intraveineuse (IV) ont une biodisponibilité de 100 % dès leur administration, alors que les médicaments administrés par voie orale ne sont biodisponibles qu'au moment où ils atteignent la circulation sanguine.
#mla_gallery-3 { margin: auto; width: 100%; } #mla_gallery-3 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-3 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-3 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
-
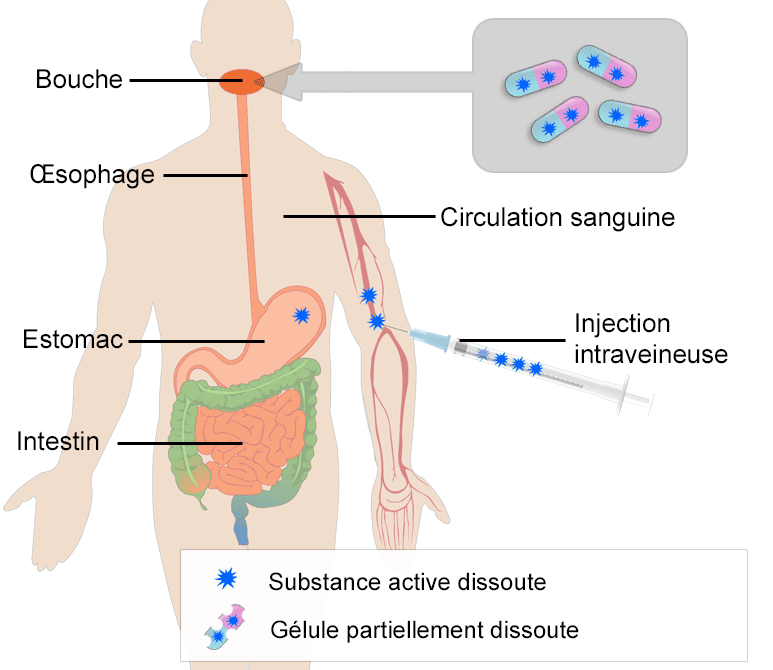
- Diagramme illustrant l’absorption d’une gélule prise par voie orale et une injection directement dans la circulation sanguine (injection intraveineuse). Après avoir atteint l’estomac, la gélule est transportée dans l’intestin grêle ou son absorption se poursuit.
Les études de biodisponibilité évaluent le taux d'absorption d'un médicament en mesurant la concentration maximale du médicament (Cmax) dans la circulation sanguine et le moment auquel cette concentration maximale est observée (Tmax). L'aire sous la courbe représente l’exposition totale de l'organisme au médicament et permet d'étudier l'étendue de l'absorption.
#mla_gallery-4 { margin: auto; width: 100%; } #mla_gallery-4 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-4 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-4 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
-
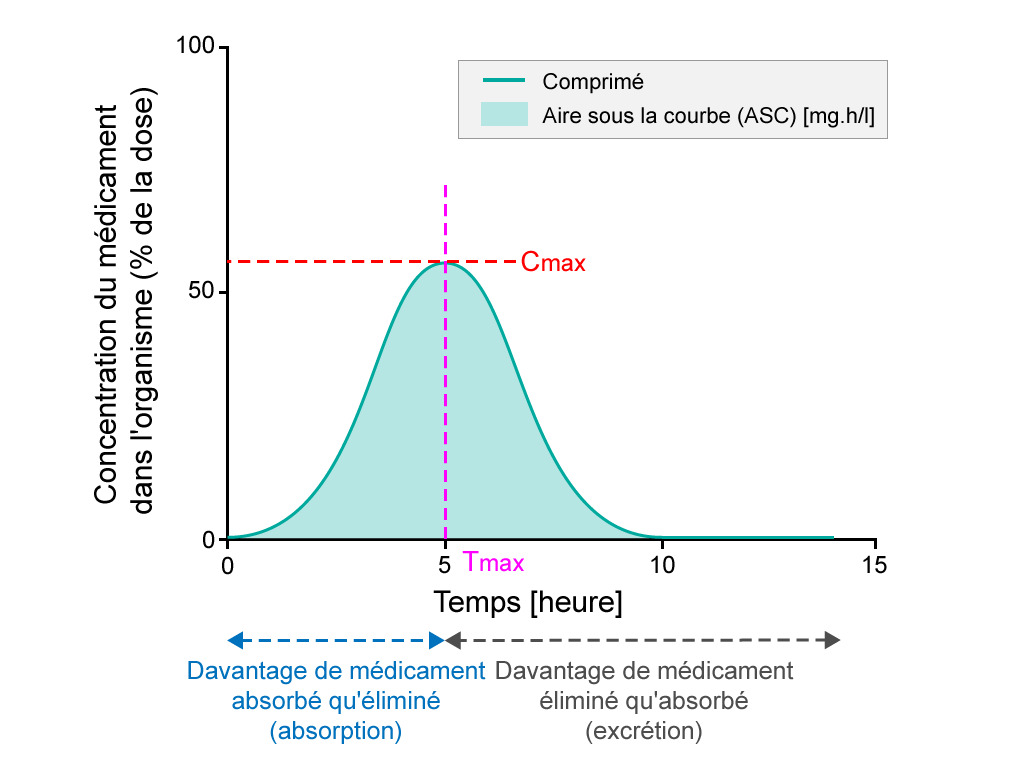
- Pourcentage de principe actif après la prise d’un comprimé, étudié sur une période de 15 heures. L’ASC est ombrée. Le Tmax est le moment où la concentration du médicament est maximale dans la circulation sanguine, tandis que la Cmax est la concentration maximum du médicament dans la circulation sanguine.
Méthodologie des études
Ces études sont habituellement conduites sous la forme d'essais croisés, randomisés et à dose unique, auprès de participants sains. Les chercheurs mesurent la concentration du médicament et ses principaux métabolites actifs dans le sang et le plasma des participants.
Études de bioéquivalence
Les études de bioéquivalence analysent la relation entre deux formulations différentes. Elles étudient également le taux et l'étendue d'absorption d'un médicament, mais en les comparant au taux et à l'étendue d'absorption d'un autre médicament ou d'une formulation différente du même médicament (formulation/médicament de référence). Les études de bioéquivalence permettent de comparer les médicaments génériques à leur médicament de référence. Un médicament doit remplir un ensemble de critères pour être considéré bioéquivalent à un autre médicament.
Études d'interaction alimentaire
Objectifs des études
Les études d'interaction alimentaire évaluent l'impact de la nourriture sur le taux, l'étendue et la biodisponibilité de l'absorption d'un médicament dans une formulation donnée. Les informations tirées des études d'interaction alimentaire sont précieuses pour les instructions d'administration figurant sur la notice, qui précisent si le médicament doit être pris à jeun ou au cours des repas.
Méthodologie des études
Ces études sont habituellement conduites sous la forme d'essais croisés à dose unique, qui comparent deux situations : des participants ayant jeuné et des participants ayant reçu un repas riche en graisses et en calories. L'étude comporte généralement deux séquences et est menée auprès de participants sains, en utilisant la concentration la plus élevée du médicament.
Études portant sur l'atteinte de la fonction rénale
Objectifs des études
Les études portant sur l'atteinte de la fonction rénale ont pour objectif d'évaluer le médicament chez des personnes présentant différents degrés d'atteinte de la fonction rénale. Ces études rassemblent des informations sur l'impact de l'insuffisance rénale sur l'excrétion des médicaments par l'organisme et sur les recommandations de dosage chez les patients présentant des degrés divers d'atteinte de la fonction rénale.
Méthodologie des études
Les études portant sur l'atteinte de la fonction rénale sont menées sous la forme d'essais à dose unique en groupes parallèles, auprès de volontaires sains de sexe masculin et de sexe féminin (environ six par groupe). Les groupes sont stratifiés par marqueurs biologiques de la fonction rénale.
Études portant sur la déficience hépatique
Objectifs des études
Les études portant sur la déficience hépatique ont pour objectif d'évaluer le médicament chez des personnes présentant différents degrés de déficience hépatique. Ces études examinent l'impact de la déficience hépatique sur les propriétés pharmacocinétiques du médicament et de ses métabolites, permettant ainsi d'établir des recommandations de dosage selon les degrés de déficience hépatique, à des fins d'efficacité et/ou de sécurité.
Méthodologie des études
Si les résultats pharmacocinétiques des études précédentes sont linéaires et indépendants du temps, les études portant sur la déficience hépatique sont généralement menées sous la forme d'essais en groupes parallèles de volontaires sains de sexe masculin et de sexe féminin (environ huit individus) présentant des degrés variés d'atteinte de la fonction hépatique. Les groupes de traitement sont stratifiés selon les classifications traditionnelles de la déficience hépatique.
Si le médicament est métabolisé par un enzyme qui n'existe qu'à la suite d'une mutation génétique, alors les participants doivent être évalués en fonction du statut de leur génotype.
Études portant sur les interactions médicamenteuses
Objectifs des études
Les études portant sur les interactions médicamenteuses évaluent l'impact d'une médication concomitante sur les propriétés pharmacocinétiques du médicament expérimental, ainsi que l'impact du médicament expérimental sur les propriétés pharmacocinétiques des médications concomitantes.
Méthodologie des études
Ces études sont guidées par les résultats in vitro précédents ; elles sont habituellement menées sous la forme d'essais croisés. Les études portant sur les interactions médicamenteuses sont menées auprès de volontaires sains ou de patients s'il est souhaitable d'évaluer les critères pharmacodynamiques en cas de médicaments trop toxiques (par exemple, les traitements anticancéreux).
Généralement, ces études se présentent sous la forme d'un essai croisé. La posologie, les intervalles posologiques, le nombre de doses, les voies d'administration et le moment de l'administration simultanée doivent viser à optimiser la détection potentielle d'une interaction, et refléter le plus fidèlement possible l'environnement clinique. Le niveau d'interaction (inhibition/induction) est classé en fonction de la modification de l'absorption de l'un des médicaments expérimentaux, et calculée comme l'aire sous la courbe de la substance.
Études QT approfondies (TQT)
Objectifs des études
Un intervalle QT mesure le rythme cardiaque. Un intervalle QT se mesure à l'aide d'un électrocardiogramme (ECG) et peut servir de marqueur biologique (imparfait) pour évaluer le risque qu'un médicament provoque une arythmie. Dans un ECG, l'activité électrique du cœur est mesurée et affichée sous forme d'ondes appelées « P », « Q », « R », « S » et « T ». L'intervalle QT correspond à la mesure entre le début de l'onde Q et la fin de l'onde T.
Méthodologie des études
Les études TQT sont réalisées in vivo pour toutes les nouvelles entités moléculaires. Elles doivent être réalisées avant les essais de phase III, quels que soient les résultats in vitro ou non cliniques.
En général, les études TQT sont conduites sous la forme d'essais croisés à dose unique auprès de participants sains. Les chercheurs évaluent les doses thérapeutiques et suprathérapeutiques du médicament par rapport à un témoin positif (un antibiotique courant comme la moxifloxacine) et un témoin négatif (un placebo).
A2-5.03.4-V1.1