Tworzenie leków. Etap 5: Badania niekliniczne dotyczące bezpieczeństwa stosowania
Wprowadzenie
Przeprowadzenie wszystkich badań i prac koniecznych do wprowadzenia nowego leku na rynek zajmuje ponad 12 lat i kosztuje średnio ponad 1 mld euro.
Odkrywanie nowych leków jest przedsięwzięciem o wysokim ryzyku. Większość (około 98%) opracowywanych substancji nie trafia na rynek w postaci nowych leków. Zwykle powodem tego jest fakt, że korzyści i ryzyko (negatywne działania niepożądane) ujawniane w procesie rozwoju nie wytrzymują porównania z lekami już dostępnymi dla pacjentów.
Opracowanie nowego leku można podzielić na 10 etapów. Ten artykuł dotyczy etapu 5: badania niekliniczne dotyczące bezpieczeństwa stosowania.
#mla_gallery-1 { margin: auto; width: 100%; } #mla_gallery-1 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-1 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-1 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
-
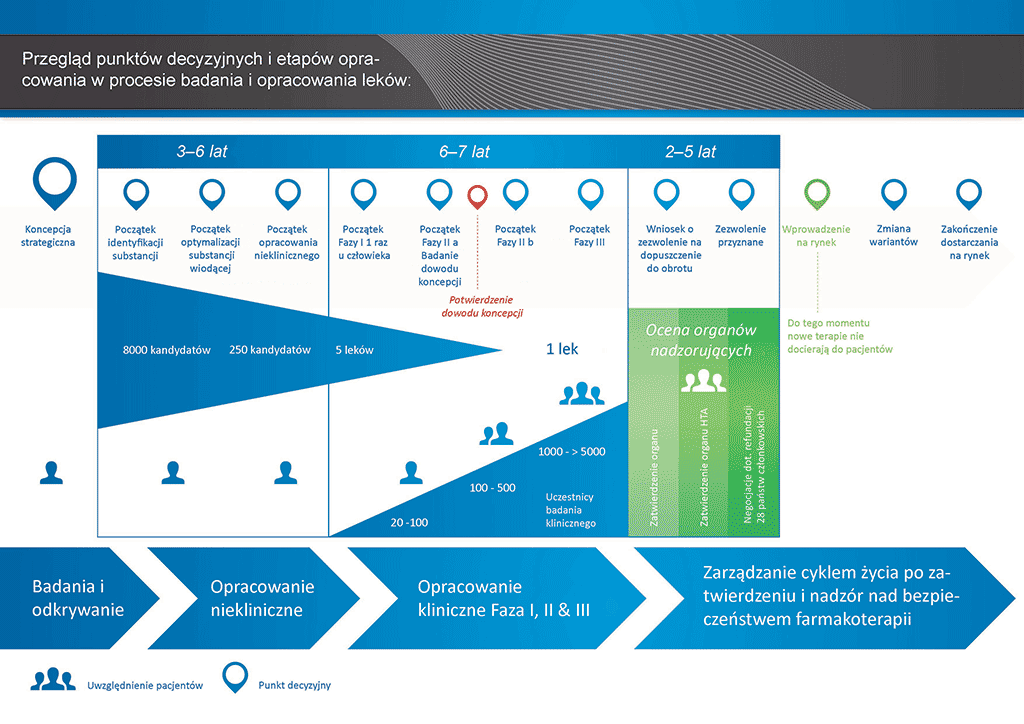
- Proces opracowania leku zajmuje ponad dziesięć lat planowania i badań pozwalających przejść od cząsteczki do dostępnego na rynku leczenia.
Etap 5: Badania niekliniczne dotyczące bezpieczeństwa stosowania
Czy przejście do badań klinicznych jest bezpieczne? Ten etap procesu rozwoju leku obejmuje testowanie bezpieczeństwa na zwierzętach, prowadzone zgodnie z regułami i przepisami Dobrej Praktyki Laboratoryjnej. Żadnego kandydata na lek nie można testować na ludziach (w „badaniach klinicznych”) przed określeniem profilu bezpieczeństwa w trakcie badań na zwierzętach. Opracowywanie leków jest ściśle kontrolowane. Istnieją zasady i przepisy prawa dotyczące tego, co i jak ma zostać wykonane.
Zanim będzie możliwe przeprowadzenie badań nieklinicznych, trzeba wytworzyć duże ilości cząsteczki „kandydującej”, aby można było wykonać odpowiednie testy. Ten proces produkcyjny także podlega rygorystycznym normom i przepisom, określanym jako Dobra Praktyka Wytwarzania.
Przepisy określają, jakie badania należy wykonać i jakie gatunki zwierząt powinny zostać wykorzystane, aby uzyskać odpowiednie informacje. Przepisy uwzględniają ocenę efektów działania:
- na zwierzę ogólnie;
- na wszystkie tkanki i narządy zwierzęcia (badania toksyczności układowej);
- na zdolność zwierząt do rozrodu i normalnego rozwoju (badania toksyczności reprodukcyjnej);
- na skórę lub oczy (badania toksyczności miejscowej);
- alergie (badania nadwrażliwości);
- na chromosomy i geny (badania genotoksyczności);
- wszelkiego wpływu na powstawanie nowotworów (badania kancerogenności).
Badania te przedstawiono poniżej.
Rodzaje badań toksyczności
- Badania toksyczności układowej
- Badania po podaniu dawki pojedynczej
- Badania po podaniu dawek wielokrotnych
- Badania toksyczności reprodukcyjnej
- Badania płodności mężczyzn
- Badania wpływu na rozrodczość i rozwój
- Badania toksyczności miejscowej
- Badania nadwrażliwości
- Badania genotoksyczności
- Badania kancerogenności (rakotwórczości)
Te badania nie tylko pozwalają określić profil bezpieczeństwa u zwierząt, ale także dostarczają ważnych informacji na temat:
- sposobu dostawania się substancji do organizmu (Absorpcji);
- Dystrybucja substancji w organizmie
- rozkładania substancji przez organizm (Metabolizmu);
- usuwania substancji z organizmu (Eliminacji).
Niekiedy określa się to skrótem „ADME”.
Wszystkie te informacje pozwalają zdecydować, czy cząsteczka „kandydująca” może zostać wykorzystana w pierwszych badaniach z udziałem ludzi (klinicznych), a jeśli tak, to w jakich dawkach.
Aby było możliwe przejście do badań klinicznych z udziałem ludzi, cząsteczka „kandydująca” musi cechować się akceptowalnym profilem bezpieczeństwa we wszystkich nieklinicznych badaniach toksyczności. Jednak nie wszystkie niekliniczne badania bezpieczeństwa zostaną ukończone. Na przykład długoterminowe badania nad kancerogennością zajmują średnio dwa lata i są kontynuowane równolegle z badaniami klinicznymi.
Piśmiennictwo
- Edwards, L., Fox, A., & Stonier, P. (Eds.). (2010). Principles and practice of pharmaceutical medicine (3rd ed.). Oxford, UK: Wiley-Blackwell.
Załączniki
#mla_gallery-2 { margin: auto; width: 100%; } #mla_gallery-2 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-2 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-2 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
- Arkusz informacyjny: odkrywanie leków
Size: 1,208,398 bytes, Format: .docx
Ten arkusz informacyjny dotyczy etapów procesu odkrywania i rozwoju leków występujących zanim związek będzie mógł zostać zastosowany w testach z udziałem ludzi — od okresu poprzedzającego odkrycie leku (gromadzenie informacji o chorobie) do nieklinicznych badań bezpieczeństwa na zwierzętach.
#mla_gallery-3 { margin: auto; width: 100%; } #mla_gallery-3 .gallery-item { float: none; margin: 1.5%; display: inline-block; text-align: center; width: 97%; } #mla_gallery-3 .gallery-item .gallery-icon img { border: 2px solid #cfcfcf; } #mla_gallery-3 .gallery-caption { margin-left: 0; vertical-align: top; } /* see mla_gallery_shortcode() in media-library-assistant/includes/class-mla-shortcode-support.php */
- Prezentacja: Podstawowe zasady dotyczące odkrywania i rozwoju leków
Size: 920,260 bytes, Format: .pptx
Podstawowe zasady dotyczące odkrywania i rozwoju leków. Przeprowadzenie wszystkich badań i prac koniecznych do wprowadzenia nowego leku na rynek zajmuje ponad 12 lat i kosztuje ponad 1 mld euro. W tej prezentacji szczegółowo przedstawiono proces od odkrycia nowego leku do wprowadzenia go na rynek oraz późniejsze działania.
A2-1.02.4-v1.1