Sviluppo non clinico: principi di base
Introduzione
La fase di sviluppo non clinico (preclinico) mira prima di tutto a individuare quale terapia candidata abbia la maggiore probabilità di successo, ne valuta la sicurezza e costruisce solide basi scientifiche prima di passare alla fase di sviluppo clinico.
Inoltre, durante questa fase, il composto candidato deve soddisfare obiettivi di tipo non medico, tra cui la definizione dei diritti di proprietà intellettuale e verificare che sia disponibile un quantitativo sufficiente di farmaco per gli studi clinici. Lo sviluppo non clinico di un farmaco è complesso e disciplinato da norme.
Fondamenti, definizioni chiave e concetti
“Non clinico” o “preclinico”?
I termini “non clinico” e “preclinico” sono spesso usati in modo intercambiabile.
Sebbene siano d’importanza cruciale nelle fasi precliniche dello sviluppo, gli studi non clinici possono essere eseguiti in qualunque momento durante il ciclo di vita del prodotto: è preferibile tuttavia che molti di essi vengano svolti prima possibile, al fine di evitare sorprese più tardi nello sviluppo.
Oltre che nell’individuazione della farmacodinamica (quello che un farmaco fa all’organismo), della farmacocinetica (quello che l’organismo fa al farmaco) e della tossicologia del candidato composto prima della somministrazione in esseri umani, i dati provenienti da studi non clinici sono utilizzati per raffinare, consolidare e aggiungere informazioni per l’aggiornamento del profilo di sicurezza del prodotto durante la fase preclinica, al momento della registrazione e durante il ciclo di vita del prodotto medicinale.
Studi in silico, in vitro e in vivo
Gli studi nello sviluppo non clinico sono condotti nelle seguenti modalità:
- In silico: “condotto su computer o tramite una simulazione al computer”, ad esempio, previsione del profilo tossicologico di un prodotto utilizzando la sua struttura chimica a partire da approcci basati su raccolte di dati;
- In vitro (latino per “nel vetro”): effettuando una procedura in un ambiente controllato all’esterno di un organismo vivente, ad esempio l’utilizzo di culture di epatociti (cellule del fegato) per studi sul metabolismo;
- In vivo (latino per “nel vivente”): esperimento che utilizza un organismo vivente intero, vale a dire animali, esseri umani o piante, invece di tessuti o cellule.
Quali sono gli aspetti fondamentali della chimica, fabbricazione e controllo (CMC, Chemistry, Manufacturing, Control) durante lo sviluppo non clinico?
Tutti gli studi di sviluppo non clinico richiedono la fabbricazione di un principio attivo idoneo:
- Per studi non clinici sono di solito necessari piccoli quantitativi (da milligrammi a grammi); un processo di “scale-up” deve essere quindi sviluppato al fine di produrne un maggior volume per gli studi clinici e più tardi, dopo l’approvazione, per il mercato
- Per studi di buona prassi di laboratorio (GLP, Good Laboratory Practice), sono richiesti lotti di principio attivo idonei o prodotti secondo linee guida di buona prassi di fabbricazione (GMP, Good Manufacturing Practice).
Alcuni passaggi fondamentali di CMC durante la fase di sviluppo non clinico comprendono:
- determinazione della dose e della somministrazione;
- caratterizzazione fisico-chimica dettagliata;
- test di stabilità e analisi delle impurità;
- metodi di sviluppo e di convalida per quantificare il principio attivo in fluidi corporei come sangue, plasma e urine in studi sull’attività e gli effetti collaterali;
- sviluppo di un prototipo ai fini dell’utilizzo in clinica.
Il processo di sviluppo non clinico
Le attività di sviluppo non clinico vanno in parallelo alle attività di ricerca. Devono supportare il programma di sviluppo pianificato affrontando gli obiettivi e le domande descritti di seguito.
Obiettivi
Una volta identificato un candidato composto, lo sviluppo non clinico deve iniziare a rispondere alle seguenti domande e le risposte verranno da valutazioni/studi specifici:
- Funziona? → valutazione dell’efficacia
- Come sarà somministrato e come reagirà l’organismo? → determinazione del profilo
- È sicuro? → tossicologia/sicurezza
- La produzione è praticabile e controllabile?
Le attività di sviluppo non clinico possono continuare per tutto il ciclo di vita del prodotto sebbene quanto prima queste domande ottengono risposta, tanto più è facile identificare il profilo del paziente che ne beneficerà maggiormente.
Gestione del progetto
Il programma di sviluppo non clinico è complesso e richiede una gestione solida del progetto e capacità comunicative nel guidare team multidisciplinari. L’equipe del progetto deve conoscere il programma clinico previsto allo scopo di definire il piano non clinico e le attività relative.
Il profilo fornisce un quadro per condurre una strategia di sviluppo non clinico, determinando gli obiettivi, rischi, responsabilità, metriche e il processo decisionale riguardo all’eventualità di procedere o meno. L’applicazione di un profilo aiuta a mantenere l’attenzione del progetto su criteri chiave del prodotto, a prendere decisioni puntuali riguardo all’eventualità di proseguire lo studio e a ridurre il suo rischio globale (vale dire, lo sviluppo continuo di un prodotto non utile).
Linee guida normative non cliniche
Nello sviluppo clinico di medicinali sono coinvolte molte parti e ogni organizzazione o istituzione segue il proprio insieme di regole. Ad esempio, le aziende hanno le loro procedure operative standard (POS). Oltre alle disposizioni di buona pratica clinica, linee guida sono consultabili sul sito web dell’Agenzia europea per i medicinali (EMA, European Medicines Agency).
- Sono generali oppure più specifiche, rivolgendosi ad aspetti scientifici e tecnici (ad esempio, specifiche per studi tossicologici necessari).
- Per qualsiasi nuova richiesta di autorizzazione all’immissione in commercio, le linee guida devono essere seguite rigidamente; eventuali difformità devono essere giustificate.
I dati sono presentati secondo il formato del documento tecnico comune (CTD, Common Technical Document) come definito dalla Conferenza internazionale sull’armonizzazione (ICH, International Conference on Harmonisation) dei requisiti tecnici per la registrazione dei prodotti farmaceutici ad uso umano. L’accordo per unire tutte le informazioni sulla qualità, sicurezza ed efficacia in questo formato comune (il CTD) ha rivoluzionato il processo di revisione normativa e ha portato a richieste armonizzate in formato elettronico che, a sua volta, consentono l’applicazione di buone pratiche di revisione. Per l’industria, ha eliminato la necessità di rimodellare le informazioni per la presentazione della domanda alle diverse autorità di regolamentazione (l’ICH riunisce le autorità di regolamentazione e l’industria farmaceutica europea, del Giappone e degli Stati Uniti per discutere aspetti scientifici e tecnici della registrazione di medicinali).
-
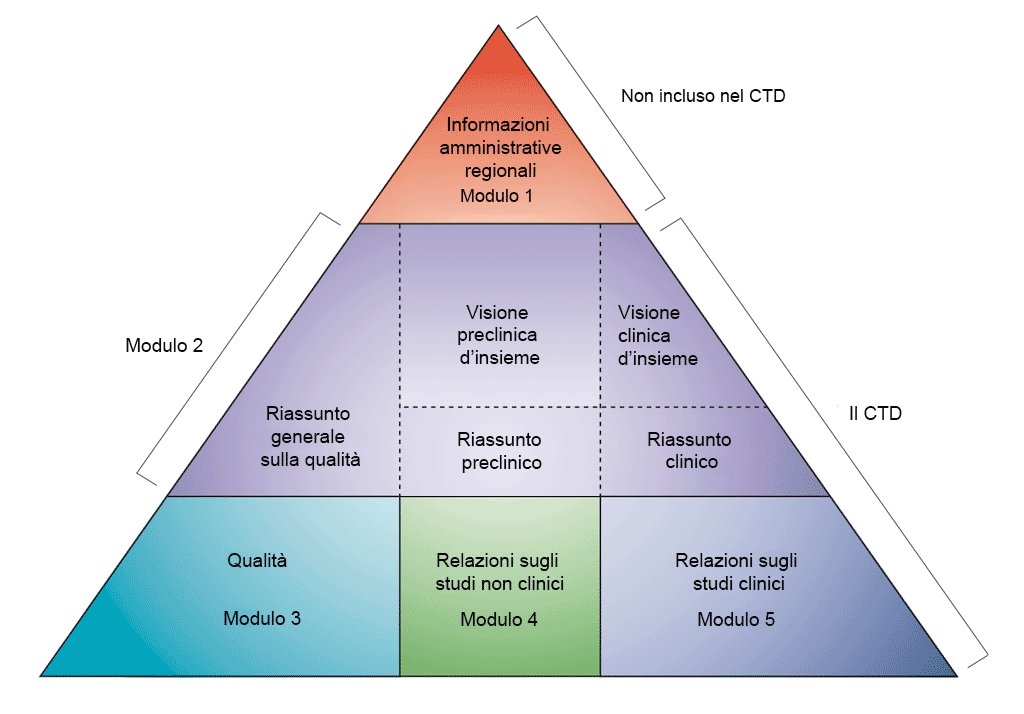
- Sviluppo non clinico nei moduli del documento tecnico comune (Common Technical Document, CTD). Adattato da ICH CTD (vedere riferimento bibliografico 1)
Il CTD è organizzato in cinque moduli (vedere la figura sotto). Nel luglio 2003, il CTD è diventato il formato obbligatorio per domande di autorizzazione all'immissione in commercio nell'UE e in Giappone e quello fortemente consigliato per le richieste relative a nuovi farmaci (NDA, New Drug Applications) presentate all'Agenzia per gli alimenti e i medicinali (FDA, Food and Drug Administration) degli Stati Uniti.
Riepilogo
La fase di sviluppo non clinico è fondamentale e deve prevedere i problemi potenziali prima che un composto passi alla fase di sviluppo clinico.
L'ammissione di un composto candidato a studi clinici richiede:
- una valutazione non clinica della sicurezza ottenuta in condizioni di buona prassi di laboratorio (GLP);
- produzione condotta con un appropriato controllo di qualità;
- dati e procedure documentati secondo il formato di CTD e basi per la fase di sviluppo clinico.
Vi è una crescente tendenza a disegnare proprietà farmaco-simili in silico e a utilizzare metodi bioinformatici per la costruzione di modelli e previsioni.
Le linee guida per lo sviluppo non clinico sono soggette a una costante armonizzazione tra le più importanti autorità di regolamentazione (Europa, Stati Uniti e Giappone). L'ICH emana regolarmente orientamenti dettagliati per l'industria farmaceutica simili a quelli pubblicati dall'agenzia europea (EMA) e statunitense (FDA).
Risorse aggiuntive
- European Medicines Agency (2007a). Guideline on strategies to identify and mitigate risks for first-in human trials with investigational medicinal products. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf
- European Medicines Agency (2007b). Guideline on requirements for first-in-man clinical trials for potential high-risk medicinal Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002989.pdf
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf
- European Medicines Agency (2011). Committee for medicinal products for human use (CHMP) ICH guidelines S6 (R1) – preclinical safety evaluation of biotechnology-derived pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002828.pdf
[glossary_exclude]Riferimenti bibliografici
- Image reproduced from ICH (2015). M4: The Common Technical Document. Retrieved 11 July, 2021, from https://www.ich.org/page/ctd
- European Medicines Agency (2009). ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. London: European Medicines Agency. Retrieved 25 June, 2015, from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002720.pdf[/glossary_exclude]